
#NephJC Chat
Tuesday April, 25, 2023 at 9 pm Eastern
Wednesday April 26, 2023, at 9 pm Indian Standard Time and 3:30 pm GMT
N Engl J Med 2023 Mar 16;388(11):969 - 979. doi:10.1056/NEJMoa2202396
Inaxaplin for Proteinuric Kidney Disease in Persons with Two APOL1 Variants
Ogo Egbuna, Brandon Zimmerman, George Manos, Anne Fortier, Madalina C Chirieac, Leslie A Dakin, David J Friedman, Kate Bramham, Kirk Campbell, Bertrand Knebelmann, Laura Barisoni, Ronald J Falk, Debbie S Gipson, Michael S Lipkowitz, Akinlolu Ojo, Mark E Bunnage, Martin R Pollak, David Altshuler, Glenn M Chertow; VX19-147-101 Study Group
PMID: 36920755




Introduction
In 2010, Genovese et al identified two common coding variants in the APOL1 gene that contribute to high rates of non-diabetic kidney disease in patients of recent West African ancestry. These risk alleles, termed G1 and G2, have minor allele frequencies of 23% and 13%, respectively, and became common among individuals of West African ancestry due to a survival advantage they confer against Trypanosoma brucei rhodesiense, a parasite that causes African sleeping sickness. Although a single copy of a G1 or G2 allele provides protection against trypanosomes, homozygosity or compound heterozygosity (G1/G1, G2/G2, or G1/G2 haplotypes) lead to a significantly higher risk of developing multiple kidney diseases, including focal segmental glomerulosclerosis (FSGS), focal global glomerulosclerosis (FGGS), HIV-associated nephropathy (HIVAN), and lupus nephritis.

Pathologic features of APOL1 nephropathy. APOL1-associated kidney disease can manifest on kidney biopsy as collapsing glomerulopathy (upper left, phenotype of HIVAN and COVAN), focal segmental glomerulosclerosis (upper right, primary finding in the majority of cases), focal global glomerulosclerosis (lower left, solidified type), and chronic changes to the renal interstitium (lower right, microcystic tubular dilation). Some or all of these may be seen in APOL1-associated kidney disease.
Multiple mechanisms have been proposed for APOL1-mediated nephrotoxicity. These include mitochondrial dysfunction and injury, impaired autophagy, pyroptosis, circulating factors, and ER stress. The leading hypothesis, however, is that podocyte-intrinsic G1 or G2 APOL1 creates pores in the cell membrane in the same way that circulating G1 or G2 APOL1 punches holes in the organelles of invading trypanosomes, as illustrated in the figure below from a recent review on mechanisms underlying APOL1 nephropathy.

Despite this level of mechanistic understanding, we do not have an APOL1-targeted therapy that is approved for clinical use. A prior pre-clinical study in mice used antisense oligonucleotides to reduce APOL1 expression to prevent the onset of kidney disease, but treatment needed to be started before the onset of detectable proteinuria. Egbuna et al’s study, discussed below, reports the in vitro and clinical effects of an APOL1 channel-blocking small molecule, inaxaplin (VX-147) (this figure).

The Study
Methods
This study combines pre-clinical validation of a small molecule inhibitor of APOL1 channels called Inaxaplin with an open-label, phase 2a clinical study evaluating the safety and efficacy of inaxaplin treatment. The pre-clinical validation work used both an in vitro assay of ion transport (thallium flux assay) and a transgenic mouse model of APOL1 nephropathy. The clinical study enrolled 16 patients in the US, UK, and France; thirteen patients had sub-nephrotic range proteinuria and 3 had nephrotic range proteinuria at the time of enrollment.
The sponsor, Vertex Pharmaceuticals, funded and designed the trial, collected and analyzed the data in conjunction with the steering committee, and helped to author the manuscript
The clinical study was divided into two parts, a required part A and an optional part B. In part A, the participants received inaxaplin treatment for 13 weeks (15 mg once daily for 2 weeks, followed by 45 mg once daily for 11 weeks) and their proteinuira was monitored. After treatment was completed at week 13, a safety visit was performed at week 17 (see figure below). In part B, participants were followed through week 25 to monitor the longer-term impact of a 13 week course of inaxaplin on proteinuria.

Study Participants:
Enrolled patients needed to have a high-risk APOL1 genotype (G1/G1, G2/G2, or G1/G2) with biopsy-proven FSGS. There was no maximum time from biopsy to enrollment.
Study participants were of 18-65 years of age, had a body weight of >50 kg and BMI 18.0-40.0 kg/m2, and needed to have >0.7 g protein/g creatinine.
Minimum estimated GFR was 27 ml/min/1.73 m2.
Maximum proteinuria was 10 g proteinuria/day.
Patients could be on immunosuppressants (tacrolimus, mycophenolate, cyclosporine, or prednisone < 10 mg daily) and RAS inhibitors, as long as doses ofwere stable for 28-days prior to enrollment.
Outcomes:
Primary outcome
The percent change from the baseline urinary protein-to-creatinine ratio at week 13. The protein-to-creatinine ratio was calculated as the mean of three early morning samples within a week time.
Secondary outcomes
The safety analysis was based on adverse events, clinical laboratory values, standard 12-lead electrocardiograms, and vital signs.
Sample size and analytic plan
The authors were unable to express heir power analysis in a way that physicians could understand and instead wrote this
Assuming a standard deviation of 0.672 (in log scale), we calculated that a sample size of 10 would provide the study with a precision of 0.294 (in log scale) for the estimation of the geometric mean percent change from baseline in the urinary protein-to-creatinine ratio.
They concluded that a sample size of 10 would provide sufficient precision for the estimation of the percent change in the urinary protein-to-creatinine ratio from baseline. The primary efficacy analysis included all the participants who had at least 80% adherence to inaxaplin treatment. Results
Results
In the pre-clinical portion of the study, inaxaplin was found to block APOL1’s ability to transport cations and to reduce the level of urinary albumin in a transgenic mouse model.
The results of pre-clinical development of Inaxaplin are summarized in the table below.
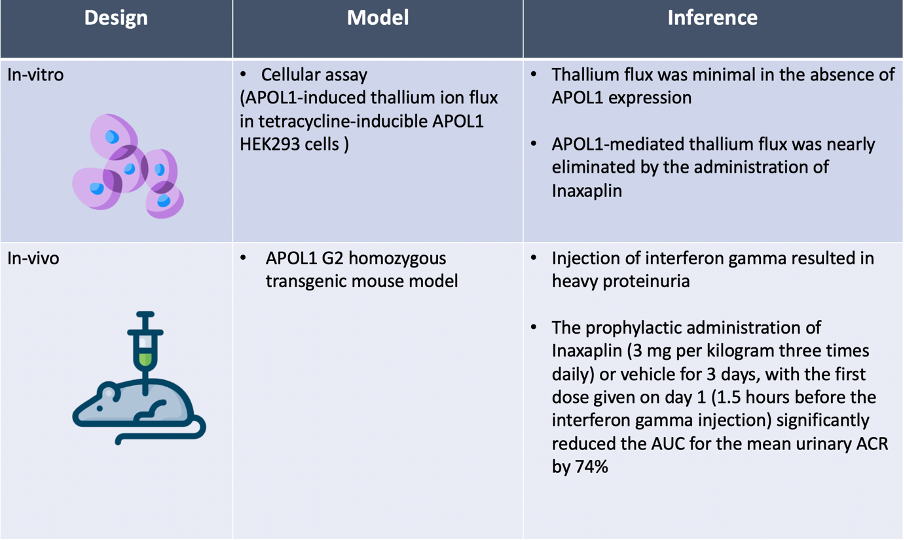
Although Jennie and Josh would gladly spend hours discussing these data, we will focus our discussion on the clinical portion of the study.
Patient cohort
A total of 58 patients were screened of whom 16 were eligible for the trial. Three patients had nephrotic range proteinuria (>2.7 UPCR) and 13 patients had subnephrotic proteinuria (0.7-2.6 UPCR). Three patients in the subnephrotic proteinuria sub-group did not adhere to the treatment protocol and were not included in the efficacy analysis. Part B of the trial was optional and was designed as an off-treatment extended follow-up for 12 weeks. Ten patients completed the optional follow-up and were included in the analysis.
The CONSORT diagram is shown below.

Study population:
The study participants had a mean age of 38.8 ± 14.5 years and the mean baseline UPCR was 2.08 ± 0.90 g/g in patients who received at least one dose of Inaxaplin. There was no difference in the 13 patients included in the efficacy analysis compared to the overall study cohort (mean baseline UPCR 2.21 ± 0.95 g/g). All patients enrolled in the study had two APOL1 risk variants. The baseline characteristics, including patient age, sex, APOL1 genotype, eGFR, distribution of standard of care medication among participants are shown in the table below.

Outcomes
Inaxaplin treatment reduced proteinuria in 12 of 13 participants. Proteinuria began declining by day 15 and continued to steadily decrease through 13 weeks of follow-up. The geometric mean change in proteinuria, as measured by the urine protein-to-creatinine ratio, was -47.6% ( 95% CI -60 to -31.3). Reduction in proteinuria was independent of the degree of proteinuria at baseline, baseline RASi use, or baseline immunosuppressive therapy use. The change of proteinuria at 13 weeks is shown in the figure below.


In part B of the study, 9 patients were followed for an additional 12 weeks off inaxaplin treatment. This was performed to determine whether the proteinuric response would be maintained following cessation of treatment. The mean UPCR increased from -47.6% to -30.1% at week 4 but remained stable thereafter (see Figure S6 below).

Safety
Thirteen patients took at least 80% of their inaxaplin doses, and were monitored for adverse safety outcomes. None of these patients discontinued treatment due to adverse events. Patients had stable blood pressure, no significant changes on electrocardiography, and preserved kidney function without a decline in GFR throughout the study. There were no safety issues with regards to biochemical parameters such as hemoglobin, bilirubin, or lipid profile. However, among 16 patients, 15 (94%) had at least one adverse event. All were mild to moderate in severity. The adverse events noted were headache, back pain, nausea, diarrhea, decrease in bicarbonate levels, diarrhea, dyspepsia and fatigue. One patient had DVT and uterine leiomyoma, neither of which were considered related to inaxaplin. A summary of the adverse events are included in the table below.

Discussion
Ten years after the APOL1 risk variants were identified, this study describes a new precision-medicine therapy that reduces proteinuria, a surrogate outcome of kidney disease. The in vitro pre-clinical work provides mechanistic evidence that inaxaplin blocks APOL1-induced ion currents. The transgenic mouse model data shows that pre-emptive administration of inaxaplin protects mice from developing progressive kidney disease. However, it is the phase 2a data showing a reduction in proteinuria in patients with genotype-validated, biopsy-proven APOL1-associated FSGS that provides the most hope for the future.
Inaxaplin was shown to be safe and well-tolerated, with no side effects that led to treatment discontinuation. Twelve out of thirteen people who received inaxaplin had a rapid and sustained reduction in proteinuria without a decrease in their eGFR; notably, this proteinuria reduction was independent of the patients’ stage of CKD or degree of fibrosis on biopsy. An exploratory analysis showed that much of this proteinuria reduction was sustained after inaxaplin treatment had been completed. Whether this proteinuria reduction will correspond with reduced rates of progressive kidney disease will require a larger, better-powered trial. The optimal timing to initiate a medicine like inaxaplin, and the optimal duration of treatment remain unknown.
Although this proof-of-concept study has many strengths, it is not without weaknesses. The open-label, single-arm design is prone to biases, including regression to the mean and confounding. The study was of short duration, and uses proteinuria as a surrogate outcome for the progression of kidney disease. It is unclear whether baseline therapy for FSGS was maximized prior to enrollment; two participants were not taking RAS inhibitors and the doses of RAS inhibitors taken by the other participants are not available. Of note, none of the patients in the trial were taking SGLT2 inhibitors. Ultimately, although the small number of participants may limit this study’s generalizability, we expect subsequent trials of inaxaplin to be larger and better-powered. (ClinicalTrials.gov NCT05312879).
Genetic testing in kidney disease in general, and in APOL1-associated kidney disease in particular, is rarely utilized. A common reason is that few genetic results result in meaningful therapeutic options. The development of genetically-targeted therapies like inaxaplin will change this discussion and encourage the use of more precision diagnostics.
Since variants in the APOL1 gene can drive multiple disease pathologies (FSGS, FGGS, HIVAN, lupus nephritis), it is possible that a single drug targeted against APOL1 function could be effective in multiple conditions. Disease-specific trials will be required to determine whether inaxaplin would reduce the rate of disease progression in these other conditions, and whether inaxaplin may modify the course of other processes in which APOL1 genotype is known to play a role (like membranous nephropathy or long-term transplant outcomes)
Conclusions
Inaxaplin is a precision medicine that targets APOL1 in focal segmental glomerulosclerosis and has been shown to reduce proteinuria in a small cohort. Larger, placebo-controlled trials with long-term follow-up are underway to discover what role this therapy will have in APOL1 associated kidney disease.
Summary prepared by
Nihal Bashir
And
Jeyakumar Meyyappan, DM
Assistant Professor, Nephrology,
Sanjay Gandhi Postgraduate Institute Of Medical Sciences
Lucknow, India
NSMC Class of 2023
Reviewed by
Jennie Lin, Tiffany Caza, Anand Chellappan, Jamie Willows, Joshua Waitzman, Joel Topf
Header image created by AI with prompts from Evan Zeitler
Do APOL1 risk variants lead to a podocytopathy? A basic science #NephJC