
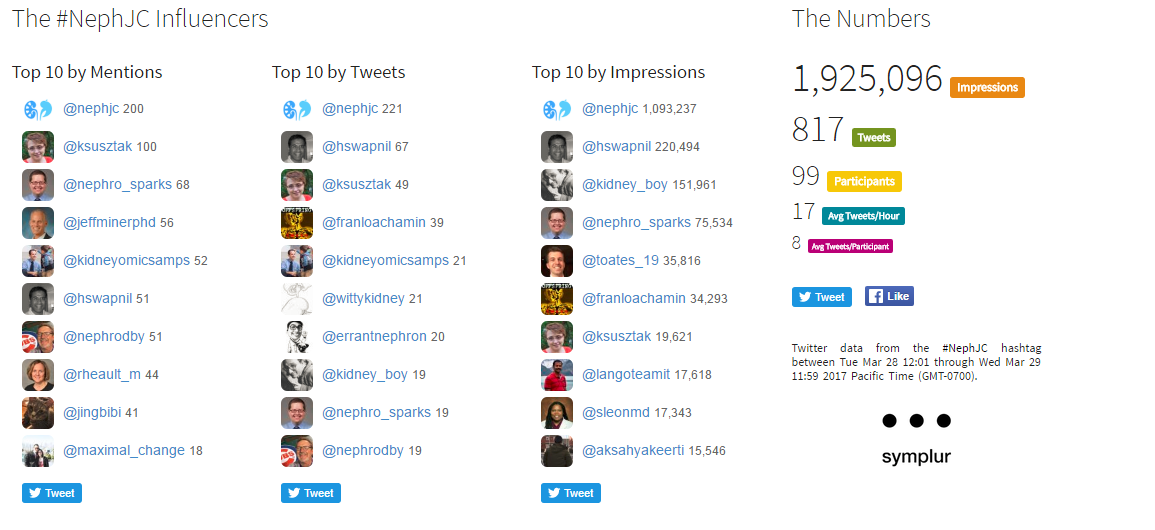
#NephJC Chat
Tuesday March 28th 9 pm EDT
Wednesday March 29th 8 pm BST; 12 noon Pacific
Nat Med. 2017 Feb 20. doi: 10.1038/nm.4287.
Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice.
Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, Boustany-Kari CM, Pullen SS, Miner JH, Hu CA, Rohacs T, Inoue K, Ishibe S, Saleem MA, Palmer MB, Cuervo AM, Kopp JB, Susztak K.
PMID: 28218918
Full Text (via ReadCube) Link


#VisualAbstract
Background
The 2010 Science paper (Genovese et al) linking variants in APOL1 with kidney disease was a massive story. In fact, it was NephMadness champion in 2015. Since its discovery we have been struggling to understand the biology of APOL1. APOL1 risk alleles are common in people of African decent. In fact, there is evidence to suggest a role in these risk alleles in the production of ApoL1 protein that help to lyse trypanosomiasis (African sleeping sickness). Thus, allowing for genetic pressure to propagate these risk alleles in areas in which trypanosomiasis is endemic. APOL1 is found on chromosome 22 in humans and some primates. However, APOL1 is not present in common animal models of kidney disease such as rats and mice. Thus it is difficult to study. Interestingly, it appears that humans can live without APOL1 and remain healthy. There are three variants of APOL1, denoted G0, G1 and G2. G1 and G2 have increased risk for kidney disease. G0 is not associated with increased kidney disease risk. It is also important to note that in order for APOL1 to enhance risk the individual must inherit 2 risk alleles. This can come in the form of 2 G1s, 2 G2s, or both G1/G2 at the same time. There are many questions that we are trying to grapple? Is APOL1 directly causing kidney disease or is this just a risk factor? Are multiple hits required? Is this a potential target for drug therapy? We are just beginning to answer these questions. Let’s quickly review what has already been published in the biomedical science literature. (Literature search conducted on PubMed on 3/23/17)
Olabisi et al. PNAS Jan 2016. in vitro model using human embryonic kidney cells
Expression of G1 or G2 let to cell swelling and cell death
Due to K efflux and activation of stress activated protein kinases (SAPKs)
Fu et al JASN Nov 2016. Drosophila model
Expression of G0 or G1 in nephrocyte causes hypertrophy and cell death
Ma et al JASN Nov 2016. in vitro model using human embryonic kidney (HEK) cells
Expression of G0, G1 or G2 in HEK cells, then gene microarray
Pathways affected include mitochondrial function, confirmed with other assays
Bruggeman et al JASN Dec 2016. Mouse model of podocyte APOL1 expression (nephrin), constitutively expressed.
Expression of G0 and G2 in podocytes of mice
Normal pathology
G2>G0 developed eclampsia, preeclampsia, small litter size
Sampson et al JASN March 2016. Human samples from NEPTUNE subjects
Increased gene expression of CXCL9, CXCL11, UBD, SNOR14B, and MUC13
Zhaorigetu et al Autophagy Nov 2008. In vitro (variety of cells)
cell death with APOL1 was autophagy dependent
Anderson et al PLoS Genetics July 2015 Zebrafish model using CRISPR-Cas9
Suppressing ApoL1 protein in zebrafish embryos results in perturbed kidney function
G1 variant appears to cause a loss of APOL1 function
G2 variant results in an altered protein that may be acting antagonistically in the presence of normal APOL1.
Methods
This paper from Beckerman et al reported in Nature Medicine represents a big step toward answering many of these questions. This group utilized a mouse model of inducible APOL1 expression only in podocytes by using a tetracycline inducible model. In this way mice are born and develop to adulthood normally until they are fed tetracycline which induces the expression of either the G0, G1, or G2 ApoL1 protein to be produced only in podocytes. See the GIF below.

Results
In order to verify if expression was truly occurring in glomeruli they imaged for GFP (as the GFP gene was also expressed in the same construct). As seen in Figure 1A GFP was observed in G0, G1, and G2 glomeruli but not WT.
In order to assess whether or not these mice developed kidney injury after the induction of podocyte specific ApoL1 G1 or G2 expression urine was assayed for albuminuria. Both G1 and G2 mice developed significant albuminuria after several weeks of doxycyline treatment (Figure 1B). Moreover they had elevated BUN and Cr (Figure 1C). Most strikingly, these mice (G1 and G2 ApoL1 overexpression in podocytes) developed sclerosis within the glomerulus (Figure 1D and below) as compared to G0 ApoL1 overexpression in pococytes.

They also look at electon microscopy and show foot process effacement in the G1/2 mice as compared to G0. They performed RNA seq (NephMadness 2017 contender) on kidney specimens of the mice and human glomeruli!! This showed pathways involved in cytokines, JAK-STAT pathway and in humans increased expression of Cxcl1, ubd, and Muc13 as prior studies have shown.
The next part of the study was to determine if the ApoL1 expression level itself was
1. cell specific
2. dose-dependent
3. reversible.
1. Cell specific- This was performed by making another mouse model in which the G0, G1, and G2 APOL1 alleles were expressed in tubular cells by using the well characterized Pax8-rtTA mice. This did not show any alterations in kidney ultrastructure, albuminuria, nor changes in BUN or Cr. Therefore, the expression of the G1 or G2 risk alleles seem to be preferentially toxic to podocytes and NOT tubular cells (Supplementary Figures 3abc).
2. Dose-dependent- In order to answer this question they correlated the the abundance of APOL1 mRNA transcript level with the amount of albuminuria. This demonstrated that amount of APOL1 transcript level correlated with the degree of albuminuria in G1 and G2 mice but not in G0 APOL1 overexpression 3-6 weeks) (Figure 2E).
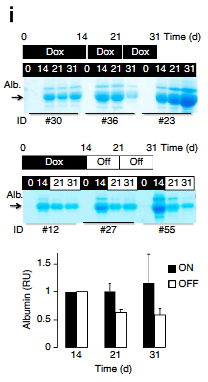
3. Reversible- In order to address this very important question the investigators treated the mice with doxycycline for 31 straight days and then another group in which the doxy was stopped after 14 days and albumin was assayed using Coomassie gels that stain all proteins in the urine. These results suggest that albuminuria can be diminished in after reduction of ApoL1 levels (Figure 2I). Does this mean that APOL1 is targetable??? Very exciting result.
So, let's quickly review. This study shows that the expression of APOL1 risk alleles G1 and G1 only in podocytes leads to albuminuria and glomerulosclerosis. Importantly, this is podocyte cell specific (at least not tubular cell), dose dependent, and potentially reversible.
What is the cellular mechanism?
This is the part of the story that typically creates the most angst for a research. Beckerman et al builds upon prior knowledge and provides compelling evidence to hammer down that it is endosomal trafficking and autophagic flux that is impaired by ApoL1. Let's take a brief journey into how they did this.
What is the Cellular Localization?
By using electron microscopy (EM) with human kidney podocytes they showed increased accumulation of intracellular late endosome/autophagosome vesicles. Using immunogold labeling and EM they show APOL1 localizing to intracellular vesicles and by immunofluorescence to the early and late endosome (EEA1 and RAB7) (Figure 3A/B). Interesting, they note that the G1 & G2 transfected HEK cells resulted in more accumulation than in G0 transfected HEK cells.
Is there a disruption in endosomal trafficking?
To explore this, they characterized the vesicles that are seen by EM in transfected HEK cells (G0, G1, & G2). This showed and increase in multivesicular bodies (MVBs) which are characteristic of late endosomes. They also found evidence of increased autophagic compartments (Figure 3g-i).
What is happening with autophagy?
Autophagy is a cellular mechanism allowing for orderly degradation of and recycling of cellular components. Prior studies have implicated autophagy in the mechanism of cell death attributed to APOL1. To examine this question they quantified the autophagic vacuoles in transfected HEK cells. This showed increased LC3II (a marker of autophagic vacuoles) in high risk G1 & G2 alleles (Figure 4a). The calculated autophagic flux by blocking the lysosomal degradation of CL3. This showed a lower degree of autophagic flux in HEK cells transfected with high risk alleles (Figure 4b). Thus, these results indicate a defect in autophagy.
What happens to the podocyte?
First, they did not detect an increase in apoptosis (Figure 5A) using staining for TUNEL staining. Therefore, they examined a type of cell death referred to as pyroptosis (inflammatory cell death). They found evidence for this in the high risk alleles by increased staining of caspase 1 (a key player in pyroptosis) and increased IL-1beta in the medium of cells expression APOL1 risk alleles (Figure 6E) and below.They tested this hypothesis of pyroptosis by treating cells transfected with high risk alleles in inhibitors of caspase 1. This resulted in diminished cytotoxicity.

Conclusion
This paper is an important advance in our knowledge about the function of APOL1 risk alleles in kidney disease. To quickly summarize the entire study.
Inducible APOL1 high risk CKD allele expression in podocytes leads (mice) leads to
Albuminuria
Glomerulosclerosis
Elevated BUN/Creat
Foot process effacement by EM
This pathology is
Cell specific (not seen in proximal tubule APOL1 risk allele expression)
at least partially reversible
Dose dependent
The mechanism of this pathology is that APOL1 risk alleles causes
abnormal endosomal trafficking and autophagic flux
enhanced podocyte cell death due to pyroptosis
This is a treasure trove of information. Of note, I purposefully only covered a few aspects of this paper and tried to hit the highlights. There is a mound of data that I reviewed but did not include in this blog post. Go to the paper to review.
The animal models are also coupled with human samples in several areas in an attempt to demonstrate the potential for these mechanisms to translate to human disease. Will this be the case? We need to start somewhere and the results presented her show that there is promise in targeting this pathway on multiple levels to diminish the impact of these risk alleles on kidney disease. How do we put these results in context with the results from Bruggeman et al that did not find significant pathology in a constitutive model of podocyte APOL1 expression. Are the levels of expression so much higher in the current study or is it the inducible nature of the above studies that leads to podocyte toxicity. Is this what would be seen in humans? Overall, this is an impressive set of studies that really take the APOL1 story to new levels. I applaud the authors on a fantastic story. I am sure we will be covering this topic alot over the next several years. Hopefully by then we will have identified a target.