

#NephJC Chat
Tuesday, October 28, 2025, 9 pm Eastern on Bluesky
Kidney Int. 2025 Oct;108(4S):S1-S71. doi: 10.1016/j.kint.2025.04.004.
KDIGO 2025 Clinical Practice Guideline for the Management of Immunoglobulin A Nephropathy (IgAN) and Immunoglobulin A Vasculitis (IgAV)
Kidney Disease: Improving Global Outcomes (KDIGO) IgAN and IgAV Work Group: Brad H Rovin, Jonathan Barratt, H Terence Cook, Irene L Noronha, Heather N Reich, Yusuke Suzuki, Sydney C W Tang, Hernán Trimarch, Jürgen Floege
PMID: 40975564




Introduction
The 2025 KDIGO Clinical Practice Guideline for the Management of Immunoglobulin A Nephropathy (IgAN) and Immunoglobulin A Vasculitis (IgAV) opens a new chapter, literally. For the first time, IgAN steps out of the 2021 Glomerular Diseases guideline to claim its own stage. (KDIGO Glomerular Diseases Work Group, Kidney Int, 2021| NephJC summary) .The timing isn’t accidental: IgAN has gone from a therapeutic drought (paradoxical given the ‘commonest GN’ moniker) to an overabundance of trials in just a few years: flozins, sparsentan, and target-release budesonide are reshaping the field faster than any consensus document can keep up.
The new guideline distils this momentum into six graded recommendations (2×1B, 4x 2B) and more than 20 practice points spanning diagnosis, risk stratification, and treatment across complex scenarios: from pregnancy to rapidly progressive disease, to pediatrics.
Proteinuria for IgAN became a validated surrogate endpoint only in 2019 (Thompson et al, CJASN 2019), yet the domino effect was immediate: a wave of accelerated drug approvals and an explosion of trial data. Compared to 2021, this edition reads less like an update but more like a snapshot of a moving target. It frames IgAN as two diseases living in one body- the immune disorder that starts it, and the chronic kidney response that tries to contain it. The result is a document that feels halfway between a guideline and a field note from the front line.
Diagnosis
Diagnosis of IgAN is most often established between the third and fourth decades of life, typically when CKD is already advanced and a significant part of the nephron mass has already been lost. This highlights the need to identify patients early and implement therapeutic strategies that can slow CKD progression.
Currently, no validated serum or urinary biomarkers exist for the diagnosis of IgA nephropathy; thus, the diagnosis is established exclusively through renal biopsy. A biopsy is indicated in patients with suspected IgAN presenting with proteinuria >0.5 g/day. Following histopathological confirmation, secondary causes should be excluded. Secondary IgA nephropathy is characterized by the deposition of IgA antibodies in the glomeruli, often associated with systemic diseases such as infections, autoimmune disorders, and liver disease. Primary IgAN is more common and associated with better outcomes than secondary, and assessment of the MEST-C score for prognosis is recommended.
Risk stratification
Risk assessment in IgA nephropathy requires combining both histologic findings and clinical disease activity rather than relying on either in isolation. The MEST-C score, remains the standard framework capturing the following components: M (mesangial hypercellularity), E (endocapillary hypercellularity), S (segmental glomerulosclerosis), T (interstitial fibrosis/tubular atrophy), and C (crescents).
Biopsy alone offers only a snapshot; KDIGO recommends the use of the International IgAN Prediction Tool, integrating clinical predictors such as proteinuria, BP control, and eGFR. (Barbour et al JAMA IM 2019; NephJC Summary| Freely Filtered episode).
Fig 1. Variables used in risk stratification tools to understand prognosis - from KDIGO guideline IgA - 2025
The International IgAN Prediction Tool merges these clinical variables with MEST-C, quantifying the risk of progression to a 50% decline or kidney failure. The tool performs well across diverse cohorts, although recalibration is important in populations with disease severity, treatment access, or demographic characteristics. Its clinical utility is discriminating patients at high or low risk (and potentially identifying those who should receive therapy).
However, a low risk of kidney failure at 5 years doesn’t mean there isn’t a lifetime risk of kidney failure (elaborated further in the “Treatment targets” section below). Since patients with IgAN classically present in the young age group, a 5-year risk for kidney failure is not representative of “long” term outcomes in IgAN but may be able to differentiate “high” risk patients, more suitable for clinical trials which mostly focus on 1-2 year outcomes.
Treatment targets
Earlier prognostic models for IgAN used proteinuria > 1 g/day as the key marker of progression. Newer data lower this to >0.5 g/day, as renal decline occurs even below the old cutoff. The treatment goal is to slow kidney function loss to <1 ml/min/year and achieve urine protein excretion <0.5 g/day, ideally under 0.3 g/day.
In the UK RaDaR cohort (Pitcher D et al, CJASN 2023, also see NephJC Summary of 2024 RaDaR data) with a median follow-up of 5.9 years, proteinuria >0.5 g/day predicted poorer renal survival (median 11.4 years with 50% reaching ESKD or death) and faster eGFR decline (-3.2 ml/min/1.73 m² per year). Even “low” proteinuria (0.44-0.88 g/day) carried a 30% 10-year kidney failure risk, versus 20% when <0.44 g/day.
The VALIGA study (Coppo R, et al. Kidney Int, 2014, N= 1147) confirmed risk with increasing time-average proteinuria: < 0.5 vs 0.5-0.9 g/day (p<0.001); 0.5-0.9 vs 1.0-1.4 g/day (p= 0.001); 1.0-1.4 g/day vs 1.5-1.9 g/day (p=0.04). Similarly, a Chinese cohort (Le W et al, NDT 2012) found a 46.5 fold higher risk of kidney failure with >1 g/day and a 9.1 fold higher risk with 0.5-1 g/day compared with <0.5 g/day.
Treatment approach
Novel therapies for IgAN have shifted the paradigm from treating hemodynamic proteinuria alone to a bi-pronged approach that also includes targeting the fundamental drivers of nephron loss.
In this context, the therapeutic approach to IgA nephropathy focuses on two fundamental objectives:
Reducing the formation of aberrant IgA molecules involved in initiating the pathogenic immune process;
Limiting nephron loss and the maladaptive mechanisms that contribute to the progressive deterioration of renal function.
Focusing solely on suppressing proteinuria can mask the glomerular injury mediated by IgA-IC complexes. Treatments in patients with more severe, rapidly progressive IgAN should include medications with anti-inflammatory and antifibrotic effects.
The “4-hit” theory represents the most widely accepted pathogenic model for IgA nephropathy, describing a complex, multistep immune process that begins with the abnormal production of galactose-deficient IgA1 (Gd-IgA1) and culminates in the glomerular deposition of immune complexes. This framework integrates genetic, mucosal, and immunologic mechanisms, providing a coherent basis for understanding disease pathogenesis and for developing targeted therapies—whether by reducing Gd-IgA1 synthesis, preventing immune complex formation, or inhibiting complement activation.
Figure 1. “Multi-hit” model hypothesis describing the pathogenesis of IgAN, from Wu MY, et al. J Clin Med, 2018.
A paradigm shift introduced by this guideline is the recommendation to initiate immunosuppressive therapy and anti-proteinuric treatment simultaneously. Traditionally, the standard approach - including that outlined in the 2021 GN guidelines - has been to start with renin–angiotensin system inhibitors (RASi) for approximately three months, and to consider immunosuppression only in cases of “insufficient response”. However, despite the fact that all randomized controlled trials referenced in this guideline implemented a 90-day period of supportive care prior to initiating immunosuppression, the current guideline proposes, as a practice point, the concomitant initiation of both generic and IgAN-specific therapies. This figure depicts the recommended guideline when approaching patients with IgAN who are considered at risk for progression.
Figure 3. Treatment targets in immunoglobulin A nephropathy (IgAN) and the positioning of drugs included in this guideline.- from KDIGO guideline IgA - 2025
Managing drivers of IgAN
The guideline gives the following two 2B recommendations regarding Nefecon and systemic steroid use:
Patients who are considered high risk should be considered for a 9-month course of Nefecon (2b). Based on the Nefigan Phase 2b (Fellström et al, Lancet 2017) and the Nefigard trials (Lafayette et al, Lancet 2023; NephJC Summary Nefigard), “Nefecon”, which is oral budesonide specifically targeted against the Peyer’s patches, is now EMA and FDA approved (as “Tarpeyo”) for use in IgAN with risk of progression. There is some debate about whether all oral budesonide formulations are equivalent, given that the guideline calls specifically for Nefecon. Formulations of oral budesonide have been used for years for IBD, it remains unclear if there are significant differences in outcomes with other appropriately dosed forms (Obrisca B, et al. Sci Rep, 2023). The data is pending on extending the course of budesonide for those who do not meet clinical remission, but the Nefigard trial showed a gradual increase in proteinuria starting from 3 months after stopping Nefecon, showcasing that IgAN may require long-term immunosuppression akin to lupus nephritis or ANCA-associated vasculitis. Unanswered questions include which patients to select for long-term therapy, and in what dose and duration to manage the risk-benefit profile in the best possible manner.
If budesonide is not available, based on the TESTING trial (Lv et al, JAMA 2022| NephJC summary), KDIGO guidelines recommend use of methylprednisolone 0.4 mg/kg/d for 2 months followed by a reduction in dose by 4 mg/d monthly for a total duration of 6-9 months (2b). This should be accompanied by prophylaxis against Pneumocystis jirovecii and antiviral prophylaxis for Hepatitis B carriers. Antimicrobial prophylaxis has been incorporated in light of the increased risk of serious infections seen in the original TESTING protocol (Lv et al, JAMA 2017).
The image below summarizes the landmark clinical trials investigating the use of systemic glucocorticoids in IgA nephropathy, showing that over the past two decades multiple randomized studies have evaluated the efficacy and safety of steroid therapy—from the Pozzi, Manno, and Lv trials demonstrating benefits on proteinuria and kidney function, to the STOP-IgAN and TESTING studies highlighting the limitations and adverse effects associated with this approach.
Image from: Landmark Nephrology- trials in IgA nephropathy
Reducing Nephron Loss
In addition to managing IgAN-specific drivers of nephron loss, it is imperative to simultaneously manage co-morbidities that accelerate eGFR loss. This includes:
1. Lifestyle measures: dietary sodium restriction (<2 g/day), smoking and vaping cessation, weight control, and endurance exercise as appropriate.
2. Blood pressure control: target ≤120/70 mm Hg (similarly to KDIGO 2021 Blood Pressure in Chronic Kidney Disease | NephJC summary)
3. Reduction of glomerular hyperfiltration and the impact of proteinuria: renin–angiotensin system blockade (RASi), dual endothelin–angiotensin receptor antagonists, or their combination with sodium–glucose cotransporter-2 inhibitors (SGLT2i).
4. Cardiovascular risk assessment and management.
5 Participation in clinical trials: all patients with IgA nephropathy should be considered for enrollment in clinical studies.
RAS inhibitors
All patients with IgAN should be treated with an optimized maximally tolerated dose of RASi (1b). This is regardless of the presence of hypertension, but should be done with caution in those with low BP, bilateral renal artery stenosis, and hyperkalemia (practice point 1.4.4.2). Use of RASi should not preclude the use of therapies targeted against IgA production or glomerular inflammation.
Until recently, treatment has focused primarily on supportive measures, particularly blockade of the renin–angiotensin system (RASi), aimed at reducing proteinuria and slowing the decline in renal function. However, a significant proportion of patients continue to experience disease progression even under optimized RASi therapy. In this context, the development and implementation of new therapies capable of slowing the progression of IgA nephropathy have become a priority, as new therapeutics become available.
Dual endothelin and angiotensin II receptor antagonists (DEARA)
Sparsentan is a dual endothelin and angiotensin II receptor antagonist (DEARA).
The recommendation for sparsentan use is supported by the findings of the PROTECT trial, a randomized, double-blind clinical study comparing the efficacy of sparsentan versus irbesartan. (Rovin BH, Lancet, 2023| NephJC summary) The study demonstrated a significantly greater reduction in proteinuria in the sparsentan group (−49.8%) compared with the irbesartan group (−15%). Moreover, sparsentan showed a beneficial effect on a hard renal endpoint, leading to a slower decline in eGFR (-6.1 ml/min/1.73 m²) compared with irbesartan (−9.9 ml/min/1.73 m²), a +3.8ml/min/1.73 m² at year 2 of the study.
Fig. 2. Percent change from baseline in urine-protein creatinine ratio in the sparsentan vs irbesartan groups at week 36 from Heerspink HJL et al, Lancet 2023
Regarding the safety profile, a higher incidence of dizziness was observed in the sparsentan group (15% vs. 6%), accompanied by an increased frequency of hypotension (13% vs. 4%). No significant differences were reported between groups with respect to serious adverse events or treatment discontinuation rates.
In addition, close monitoring of female patients receiving sparsentan is recommended due to the associated risk of fetal toxicity. Consequently, effective contraceptive methods should be used both during treatment and for at least one month post-drug discontinuation. Regular pregnancy testing is advised throughout the course of therapy. Finally, the FDA recommends monitoring liver function every month for the first one year and then 3 monthly during treatment. This is not a requirement in Europe.
Meanwhile, although not published at the time of data analysis for the guideline, more evidence favoring endothelin receptor antagonism is now available from the ALIGN trial (Heerspink et al, NEJM 2024). This trial randomized 370 patients with IgAN to atrasentan versus placebo. The prespecified interim analysis at 36 weeks demonstrated a 36% geometric mean difference in urine protein creatinine ratio relative to baseline in the atrasentan arm vs the placebo arm. The final analysis, including GFR slope data, are awaited.
SGLT2 inhibitors
Flozins, as we all very well know, act by reducing tubular reabsorption of glucose and sodium, exerting beneficial effects on the kidney that are independent of glycemic control.
Evidence supporting the use of SGLT2 inhibitors in IgAN primarily comes from the EMPA-KIDNEY (n=118 IgAN cases) and DAPA-CKD trials (n=270 IgAN cases), which included subgroups of patients with biopsy-proven IgAN. In these studies, treatment with empagliflozin and dapagliflozin led to a significant reduction in the risk of kidney disease progression and a decrease in proteinuria, even among patients without diabetes.
Adapted table s8. SGLT2i trials’ outcome, from KDIGO guideline IgA - 2025
However, participants with IgAN in these trials generally had long-standing disease and lower baseline eGFR, creating some uncertainty regarding the benefit of SGLT2 inhibitors in younger patients with relatively preserved kidney function (eGFR >60 ml/min/1.73 m²). Consequently, flozination is recommended particularly for patients with a high risk of progression and persistent proteinuria, as an adjunct to RASi, with careful monitoring for adverse events, especially genital mycotic infections. Moreover, the combination of SGLT2 inhibitors with corticosteroid therapy in IgA nephropathy appears promising for improving proteinuria control; however, it remains a matter of concern given the potentially increased risk of serious infections previously observed in the STOP-IgAN (Rauen T, NEJM, 2015) and TESTING trials (Lv et al, JAMA 2017).
Special situations
IgAN with nephrotic syndrome
This presentation is rare but may be associated with a podocytopathy resembling minimal change disease (MCD-like variant). In such cases, management should follow MCD treatment guidelines. When biopsy findings show mesangioproliferative features, patients should be managed as those at high risk for progressive renal function loss due to IgAN. Nephrotic-range proteinuria without nephrotic syndrome often reflects secondary FSGS (e.g., obesity, uncontrolled hypertension) or advanced glomerulosclerosis and interstitial fibrosis.
IgAN with acute kidney injury (AKI)
AKI can occur during episodes of gross hematuria, typically associated with upper respiratory infections. Initial management is supportive, and a repeat kidney biopsy should be considered if kidney function fails to improve within two weeks after resolution of hematuria. AKI may also result from rapidly progressive glomerulonephritis (RPGN) with extensive crescent formation, requiring urgent biopsy after excluding other causes such as ANCA-associated vasculitis, anti-GBM disease, or drug toxicity.
Rapidly progressive IgAN
Defined as a ≥50% decline in eGFR within ≤3 months, after exclusion of reversible causes. Biopsy typically shows mesangial and endocapillary hypercellularity with numerous crescents. Treatment consists of cyclophosphamide combined with systemic glucocorticoids, following the KDIGO guideline for ANCA-associated vasculitis. There is insufficient evidence to support rituximab use in this setting.
IgAN and pregnancy
Women of childbearing potential should receive preconception counseling, including discontinuation of RASi, SGLT2 inhibitors, sparsentan, Nefecon..The use of glucocorticoids in the first trimester of pregnancy is associated with a possible slight increase in the risk of cleft lip and/or palate in the fetus, although the evidence is inconclusive. However, no increased risk has been demonstrated for other major pregnancy complications such as preterm birth, low birth weight, or preeclampsia. Blood pressure should be optimized with pregnancy-safe antihypertensives. Glucocorticoid use in the first trimester may slightly increase the risk of cleft lip/palate, although data are inconsistent, and no significant associations have been found with preterm birth or preeclampsia. Nefecon is not recommended during pregnancy; budesonide appears relatively safe based on data from inflammatory bowel disease, but carries an FDA Pregnancy Category C designation.
IgAN in children
Children often present with visible hematuria and lower proteinuria than adults. Management is based on RAS blockade, salt restriction, and blood pressure control. Glucocorticoids are used in those with persistent proteinuria or high-risk features, sometimes combined with cyclophosphamide in severe cases. Clinical trial enrollment or additional immunosuppressants may be considered for non-responders. Long-term follow-up is essential, as relapses can occur years later.
Immunoglobulin A vasculitis
Definition and context
IgA vasculitis (IgAV), formerly known as Henoch–Schönlein purpura, is a small-vessel vasculitis characterized by IgA deposition, typically affecting the skin, joints, gastrointestinal tract, and kidneys.
Diagnosis
A kidney biopsy is mandatory in patients with persistent proteinuria ≥0.5 g/day for more than 4 weeks, impaired kidney function, or suspicion of rapidly progressive glomerulonephritis (RPGN).
Prognosis
Unfavorable prognostic factors include uncontrolled hypertension and the level of proteinuria at presentation and during follow-up. The Oxford MEST-C score may assist in histologic assessment, particularly the E1 component (endocapillary hypercellularity), which is associated with faster disease progression.
Treatment
Prevention of nephritis
Systemic glucocorticoids are not recommended for preventing renal involvement in patients with IgAV without nephritis.
IgAVN without RPGN
There are no proven therapies that prevent the formation of pathogenic IgA immune complexes. Management follows similar principles to those used in IgAN: blood pressure control, renin–angiotensin system blockade (RASi), possible addition of SGLT2 inhibitors, and lifestyle modification. Glucocorticoids may be used in selected cases after careful evaluation of the risk–benefit profile, preferably at low doses and with appropriate infection prophylaxis.
IgAV with RPGN
Treatment should follow the KDIGO 2024 recommendations for ANCA-associated vasculitis, consisting of systemic glucocorticoids in combination with cyclophosphamide.
Areas for future research
Discussion
These are the first-ever fully dedicated guidelines to address IgAN and IgAV. They aim to deliver a practical, risk-aligned, and updated treatment guidance that protects kidney health, with clear direction on the roles of supportive care and immunosuppression. It advances precision risk stratification by histology, proteinuria trajectories, and validated prediction tools to better classify risk and individualize follow-up.
Classical teaching in IgAN has always been optimised supportive care before immunosuppression trials, that too only in cases which are at “high” risk of progression of disease. The current KDIGO guidelines recognise the risk of progression of disease in all patients with IgAN, including those with traditionally “low” proteinuria. RAS inhibition is now recommended in all IgAN (in contrast to those with >0.5 g/d proteinuria in previous guidelines). Supportive therapy has been expanded to include the universally loved champion of CKM syndrome, aka flozins and dual endothelin angiotensin receptor antagonists, where available.
Recognition of the risk of progression of disease in all patients with IgAN will possibly expand the use of our armamentarium of drugs, both supportive and immunosuppressive, to a wider population of IgAN cases, and possibly to all of them.
The current guideline suggests that immunosuppression becomes relevant WITH supportive therapy, and not just AFTER full optimisation, although most clinical trials and regulatory approvals of IgAN are based on a baseline optimised supportive therapy.
The workgroup recognizes two distinct pathways to target therapies to, and in that case, doesn’t it seem obvious that the major driver of IgAN should be targeted right from the word go? The main hesitation in giving immunosuppression in IgAN has been the high risk of infections- with Nefecon (TRF budesonide), it has been shown that focused targeting is possible without systemic immunosuppression. Simultaneous initiation of therapy targeting both pathways may very well become standard of care in the future.
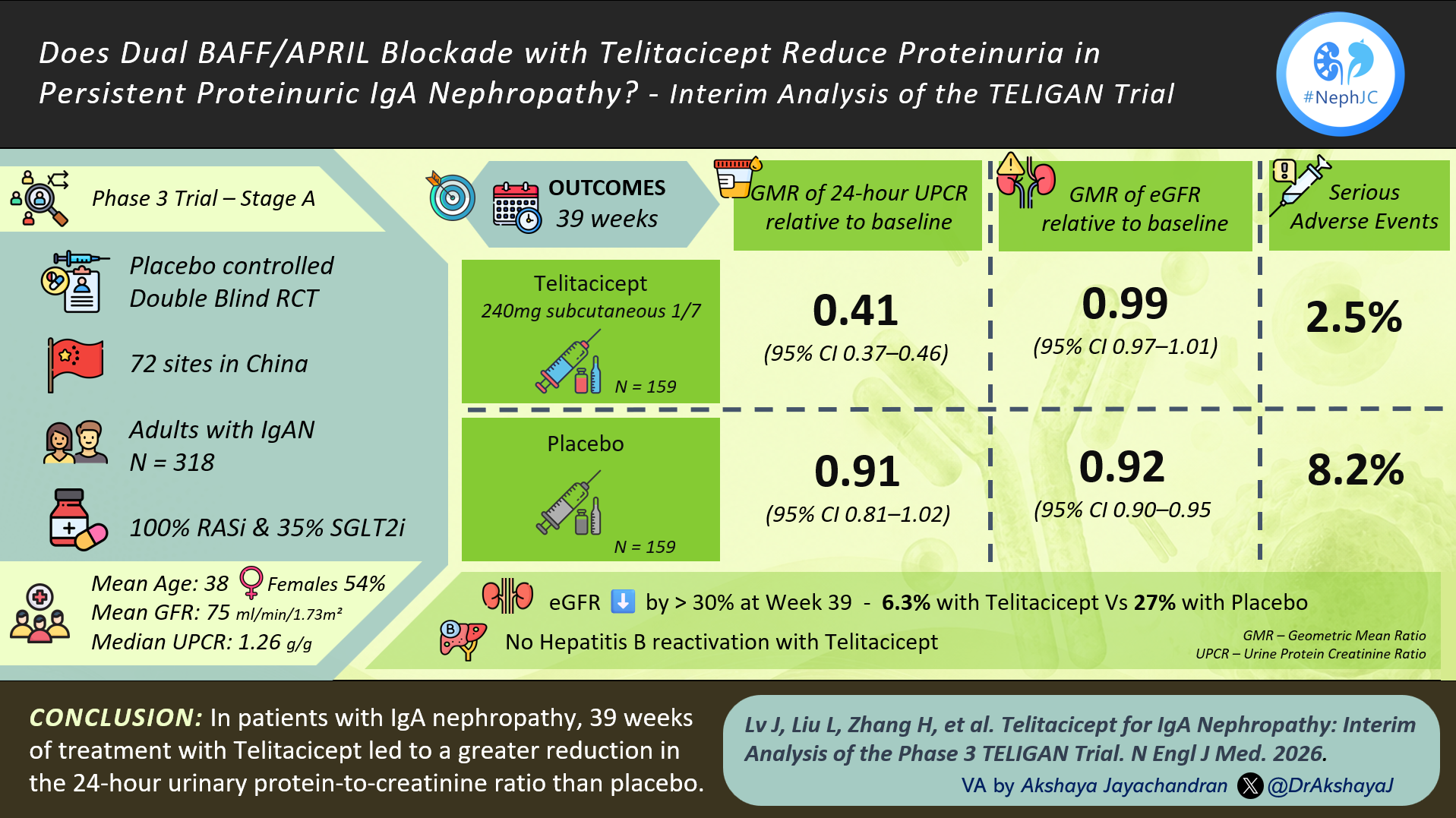
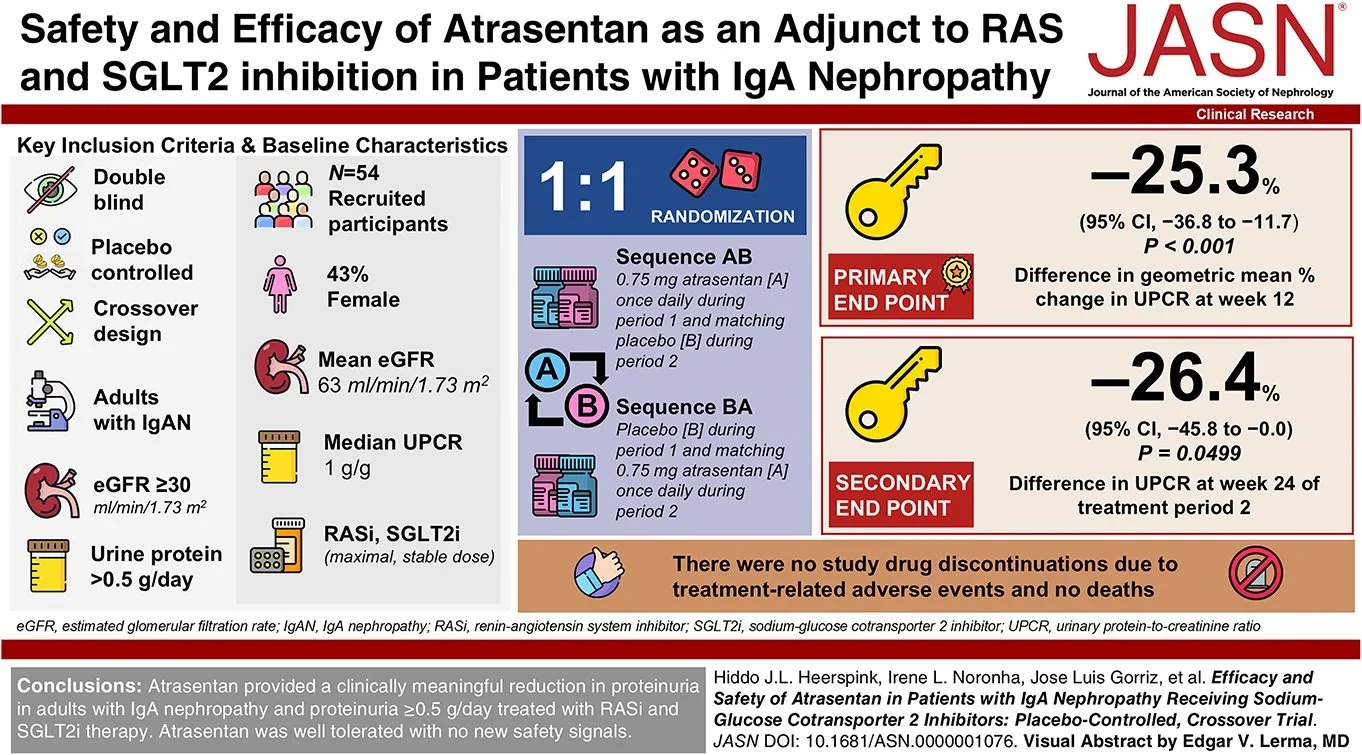
By the time the guideline was published, several major trials had already expanded the therapeutic landscape, indicating that clinical practice will soon need to evolve beyond the evidence base on which this guideline was built. Newer promising therapies with evidence of a significant decrease in proteinuria in IgAN include iptacopan (APPLAUSE IgAN trial: Perkovic et al, NEJM 2025), atrasentan (ALIGN trial: Heerspink et al, NEJM 2024), and sibeprenlimab (VISIONARY trial). Additional studies of endothelin antagonists, complement inhibition (iptacopan, avacopan, ravulizumab, IONIS-FB-LRx), and APRIL/BAFF inhibition (atacicept, telitacicept, povetacicept, sibeprenlimab) are currently underway - with many results to be presented and published in the upcoming Kidney Week. The growing number of novel immunomodulatory therapies introduces uncertainty regarding the optimal sequencing and combination of available agents.


Meaningful progress toward personalized therapy in IgAN will depend on identifying biomarkers that can guide which patients are most likely to benefit from complement inhibition, APRIL blockade, or targeted intestinal therapy with Nefecon. KDIGO highlights the complexity of this decision: patients differ in age, comorbidities, steroid tolerance, and access to therapies. Real-world experience, ongoing clinical trials, and global collaboration will be essential to ensure that advances reach all patients with IgAN, not only those in specialized centers. Ultimately, the goal remains simple but ambitious: delay and prevent kidney failure across an entire lifetime, while minimizing treatment burden and toxicity.
Conclusion
KDIGO 2025 marks a critical transition in the care of IgA nephropathy. The focus has shifted toward risk-based patient stratification, approved therapies, and treatment response. New agents and evolving evidence will continue to reshape clinical practice, and guidelines must adapt alongside them. The challenge now is to ensure that these advances reach patients worldwide and that scientific progress translates into meaningfully improved long-term outcomes for everyone living with IgAN.