

The first 2015 NephJC chat was held on
- January 6th at 9 pm EST for the Americas
- January 7th at 8pm GMT for Europe and Africa
We discussed the physiology series from CJASN, with a focus on the latest article from the series, the distal convoluted tubule.
Summary by JM Topf
Distal convoluted tubule
PMID: 24855283
The paradox of nephrology is that though we are supposed to be experts on kidney physiology, we spend most of our time taking care of patients without normal (or any) kidney function. Thus, nephrologists learn renal physiology in medical school but have surprisingly little need to update that knowledge in day-to-day rough tumble world of modern nephrology work, comprising as it does, of looking after patients with chronic kidney disease and dialysis. However, that does not give us a pass on learning the physiology. Nephrologists should not lose sight of the scientific basis of what we do, and having a sophisticated model of how the kidney works is part of the nobility of nephrology.
To help nephrologists gain an uptodate and state-of-the-art understanding of renal physiology, Melanie Hoenig, Mark Zeidel, and Paul Palevsky are editing a series in CJASN covering renal physiology (editorial describing their mission). So far the series has been anatomic, but I understand that after completing the nephron there will be further articles following a different axis. To the credit of CJASN they are making the articles open access.
The articles so far:
- Homeostasis, the Milieu Intérieur, and the Wisdom of the Nephron (PDF)
- The Glomerulus: The Sphere of Influence (PDF)
- Proximal Tubule Function and Response to Acidosis (PDF)
- Urine-Concentrating Mechanism in the Inner Medulla: Function of the Thin Limbs of the Loops of Henle (PDF)
- Thick Ascending Limb of the Loop of Henle (PDF)
- Distal Convoluted Tubule (PDF)
The latest one is the subject for our next #NephJC discussion, slated for January 6th for the Americas and January 7th for Europe and Africa.
A walk through of the article
Anatomy
The nomenclature, or more about the anatomy of the distal nephron than you ever wanted to know.

Beautiful art work. I can't overestimate the importance of good visuals.
Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014;9(12):2147-63. (Link)
The distal nephron begins just after the the macula densa cells of the thick ascending limb of the loop of Henle and continues until the tubules connect with another nephron indicating the start of the collecting ducts.
The distal nephron is made of two segments, the distal convoluted tubule and the connecting tubule.
The DCT is the shortest segment of the kidney, only 5 mm and it is further divided into two functional units:
- DCT1 or early DCT
- DCT2 or late DCT
The two units of the DCT are differentiated by their responsiveness to aldosterone. Both DCTs have mineralocorticoid receptors, but only DCT2 expresses 11-beta hydroxysteroid dehydrogenase 2 to inactivate cortisol. 11-beta hydroxysteroid dehydrogenase 2 is also expressed in the connecting tubule and cortical collecting duct.

The cortisol can activate the same mineralocorticoid receptor tissue as aldosterone. In DCT1 (and much of the body) cortisol which circulates at 1,000 times the concentration of aldosterone swamps the MR receptor and makes it essentially unresponsive to changes in the aldosterone concentration. 11beta-hydroxysteroid dehydrogenase inactivates cortisol. This makes the tissue sensitive to changes in aldosterone levels. From the Potassium and Metabolic Alkalosis Lecture found here.
Sodium
The DCT is the most mitochondria rich segment of the nephron. These mitochondria provide the ATP for reabsorbing 5-10% of the filtered sodium load, chiefly via the thiazide sensitive Na-Cl cotransporter (NCC).
A loss of function mutation in the gene that codes for NCC causes Gitelman syndrome.
The NCC is seen throughout the DCT, but the DCT2 also expresses the ENaC. Sodium flows down its concentration gradient into the cells. The movement of the cation out of the tubule, makes the tubule electronegative and drives (paracellular) chloride reabsorption and potassium secretion. ENaC is found in increasing frequency as one moves from the DCT2 to the connecting tubule to the collecting duct and so the tubule becomes more and more electrically negative.

From the Potassium and Metabolic Alkalosis Lecture found here.
In order to maintain the net movement of sodium across the tubule, Na-K-ATPase runs at a high rate. Potassium is recycled through Kir4.1 and other outward potassium channels that exist on the basolateral membrane. Defects in the potassium channel have been identified and patients present with a Gitelman-like phenotype, variously called EAST or SeSAME.
Chloride
Chloride reabsorption occurs by two means. Early in the DCT, chloride is absorbed by the tubule via the NCC. The chloride then exits the basolateral side via a chloride channel, ClC-Kb (with an accessory subunit, Barttin) or a potassium-chloride cotransporter. Since the Na-K ATPase pumps 2 potassiums in while moving 3 sodiums, it leaves a net negative charge across the basolateral membrane which helps drive chloride excretion.
Regulation
NCC activity is regulated largely by sodium delivery. As sodium delivery goes up so does the activity of NCC. This is seen with chronic loop diuretic use which causes hypertrophy of the DCT as NCC activity is upregulated.
To increase the number and activity of the NCC, two processes occur:
- Mobilization of pre-made NCC from a pool of storage vesicles inside the cells to the apical membrane
- phosphorylation of NCC by either SPAK or OSR1
WNK is an enzyme that activates SPAK and OSR1 so one could consider WNK a primary regulator of NCC, once removed.
An activating mutation of WNK1 and WNK4 cause over activity of NCC resulting in:
- Hypertension from the excessive sodium reabsorption
- Hyperkalemia from decreased distal delivery of sodium, needed by the principal cells to secrete potassium
- Metabolic acidosis from decreased distal delivery of sodium, needed by the intercalated cells to secrete hydrogen
- Hypocalciuria
This activating WNK mutation is called Familial Hyperkalemic Hypertension, or Gordon's Syndrome, or pseudohypoaldosteronism type 2

From the Potassium and Metabolic Alkalosis Lecture found here.
One of the defining characteristics of Gordon's syndrome, and a key to identifying it as a DCT abnormality, is its exquisite sensitivity to thiazide diuretics. WNK1 and WNK4 were the first mutations associated with Gordon's syndrome, but most patients with Gordon's syndrome have mutations with CUL3 and KLHL3 which are responsible for WNK degradation.
In vivo NCC is stimulated (via WNK) by aldosterone, angiotensin 2, insulin and vasopressin.

Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014;9(12):2147-63. (Link)
Calcineurin Inhibitors
Cyclosporine and Tacrolimus causes a Gordon's syndrome-like picture: hypertension, hyperkalemia metabolic acidosis and hypercalciuria. There is evidence that tacrolimus stimulates WNK-induced up regulation of NCC. This means that thiazides might be the ideal drug for the treatment of CNI-induced hypertension.
Potassium
Potassium secretion in the DCT increases as you move down the DCT. This parallels the increased electonegativity of the tubular fluid (due to sodium reabsorption through the eNaC) as you move distally. This electrogenic movement of potassium flows through the renal outer medullary potassium channel (ROMK).

Potassium secretion is also flow dependent, and that is via the maxi-K channel, or big K channel. The increased flow causes shear stress which activates the channel. (How cool is that?)
There is an interesting paragraph that proposes that the cortical collecting duct has historically been the segment most associated with potassium secretion due to the fact that it is more easily isolated than the DCT. Sometimes facts are just a side effect of experimental convenience.
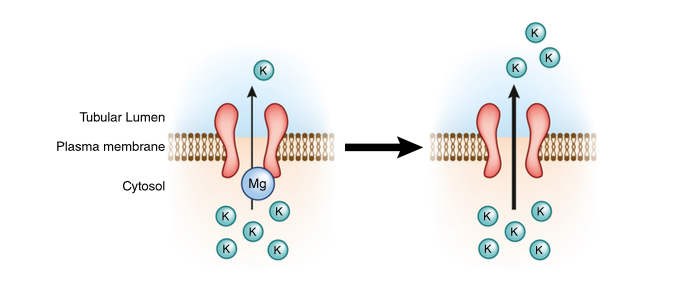
ROMK activity is enhanced by hypomagnesemia which may be the reason that hypomagnesemia is associated with refractory hypokalemia.
Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014;9(12):2147-63. (Link)
The authors discuss a phenomenon whereby increased potassium intake has a direct effect on the DCT by increasing the number of ROMK channels prior to a change in aldosterone levels. They describe similar changes in the big potassium channels, eNaC and the Na-K ATPase.
Calcium
Calcium reabsorption in the DCT occurs via the transcellular route as compared to the paracellular route in other parts of the nephron. Calcium enters the DCT cell via the TRPV5 channel and then is excreted from the basolateral membrane via the a calcium ATPase and Type 1 sodium calcium exchanger.
PTH increases TRPV5 by increasing transcription and decreasing metabolism. PTH also increases TRPV5 activity. Klotho also increases calcium reabsorption via TRPV5. Interestingly, CKD is associated with decreased Klotho activity. This maybe an important part of the abnormal calcium metabolism found in CKD.

Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014;9(12):2147-63. (Link)
The decreased urinary calcium with thiazide diuretics surprisingly has nothing to do with TRPV5 or DCT calcium handling. The hypocalciuria is due to increased proximal tubule sodium-calcium absorption from the volume deficiency induced from the thiazide diuretic. Loop diuretics cause volume deficiency, but do not cause hypocalciuria because they also indirectly stop paracellular calcium reabsorption in the thick ascending limb of the loop of Henle.
Magnesium
The DCT is the primary site of active, transcellular magnesium reabsorption (like calcium, most magnesium reabsorption occurs in the thick ascending limb of the loop of Henle, via a paracellular route). Magnesium crosses the apical membrane via TRPM6. The channel on the basolateral side has not been established though that didn't stop the authors from speculating.
Thiazides sharply down regulate TRMP6, decreasing magnesium reabsorption.

Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014;9(12):2147-63. (Link)