
#NephJC Chat
Tuesday Feb 12 at 9 pm Eastern
Wednesday Feb 13 at 8 pm GMT, 12 noon Pacific
Wednesday Feb 13 at 9 pm IST
N Engl J Med. 2019 Jan 10;380(2):142-151. doi: 10.1056/NEJMoa1806891. Epub 2018 Dec 26.
Diagnostic Utility of Exome Sequencing for Kidney Disease.
Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, Li Y, Zhang J, Nestor J, Krithivasan P, Lam WY, Mitrotti A, Piva S, Kil BH, Chatterjee D, Reingold R, Bradbury D, DiVecchia M, Snyder H, Mu X, Mehl K, Balderes O, Fasel DA, Weng C, Radhakrishnan J, Canetta P, Appel GB, Bomback AS, Ahn W, Uy NS, Alam S, Cohen DJ, Crew RJ, Dube GK, Rao MK, Kamalakaran S, Copeland B, Ren Z, Bridgers J, Malone CD, Mebane CM, Dagaonkar N, Fellström BC, Haefliger C, Mohan S, Sanna-Cherchi S, Kiryluk K, Fleckner J, March R, Platt A, Goldstein DB, Gharavi AG.
PMID: 30586318



Introduction
Since genetics play a role in the observed heritability of renal traits such as glomerular filtration rate and end-stage kidney disease (ESKD), identifying causal DNA variants (differences in nucleotides when compared to the reference genome) that promote kidney disease is a research priority.
Identification of these variants is improved with the advent of next-generation sequencing (NGS) technology, which allows unbiased detection of novel DNA variants in a high-throughput fashion. For this week’s NephJC chats, we are discussing the latest work from Dr. Ali Gharavi’s group, demonstrating how NGS, and more specifically exome sequencing, yields a genetic diagnoses in 10% of a large chronic kidney disease (CKD) cohort and reclassified the diagnosis for some of these patients.
Background on kidney genetics
Genetic testing is increasingly being used to inform the clinical management of select nephropathies. Because genetic diseases of the kidney and urinary tract are not usually caused by a single mutation across patients, genetic testing can be complex. The authors of the present paper have reviewed the various techniques used for clinical genetic testing. NGS methods such as exome sequencing (reading unbiased sequences from protein-coding portions of the human genome) are increasingly being used clinical settings due to their ability to flag uncataloged DNA variants, increased time efficiency, and relatively modest cost.
NGS is a massively parallel technology (i.e., it has the ability to read many nucleotide bases at the same time) and can be applied to many different types of biological sequencing applications, including RNA sequencing, whole exome sequencing (WES), and whole genome sequencing (WGS). For additional background on how NGS see:
An FAQ from the national library of medicine on WES
A beginner’s guide to NGS
Another detailed guide to NGS (PDF at link)
The #Nephmadness write up (from 2015!) by Conall O'Seaghdha
Or just watch this video:
After sequencing is performed, the sequencing reads are aligned to the human genome, and differences between a patient’s reads and the reference genome are candidates for disease-causing variants. DNA sequence variation might occur throughout the genome and include single-nucleotide polymorphisms (SNPs), small insertions or deletions involving <5–10 base pairs, and structural variants. Although the bioinformatics pipeline for these types of challenging analyses requires stringent quality control, the potential clinical payoff is large. For example, patients with unclear clinical diagnoses can have the precise molecular cause of their disease identified and individualized therapeutic plans with disease surveillance formed. Indeed, Dr. Gharavi’s group had recently shown in a proof-of-principle pilot study that the PARN gene is a novel locus for renal fibrosis, based on WES data of two patients. For more on the conversation surrounding integration of sequencing into clinical practice, check out these reviews.
Study design
In the present study, exome sequencing was performed on 3,315 probands (patients with disease and variants). Relatives of the probands were not sequenced.
1,128 participants were from the multi-center AURORA (A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Hemodialysis: An Assessment of Survival and Cardiovascular Events) clinical trial based primarily out of European countries. Inclusion criteria for AURORA were:
Age 50-80 and all-cause ESRD on hemodialysis for at least 3 months.
Exclusion criteria for AURORA included
Statin therapy in the past 6 months
Expected kidney transplant within one year
Other major co-morbidities (except for diabetes) limiting life-expectancy to less than one year, history of malignancy
ALT levels 3 times the upper limit of normal, history of statin-induced myopathy, uncontrolled hypothyroidism, unexplained CK more than 3 times the upper limit of normal, and serious hypersensitivity reactions to statins
The remaining 2,187 participants were from the Columbia University Medical Center (CUMC) Genetic Studies of Chronic Kidney Disease biobanking cohort. Inclusion criteria included
A clinical diagnosis of CKD, defined as the following: renal failure requiring dialysis or transplantation, serum creatinine > 1.5 mg/dL in men or > 1.3 in women
Or the presence of severe proteinuria or hematuria consistent with active glomerular disease
No exclusion criteria were noted
Genomic DNA from participants was mostly from peripheral blood, and sequences were captured and underwent paired-end sequencing for a target coverage of 111x. Sequencing coverage refers to sequencing “depth” or how many times a particular nucleotide is included in the dataset. Another way of thinking of it is that 50 million base pairs for the human exome (which is around 1.5% of the human genome) would generate a dataset of 5 trillion bases. This high depth of coverage increases confidence in variant calling because errors occur randomly during the sequencing process. The investigators then focused their analyses on 625 genes they manually curated, which can be found here. Variants were classified according to the American College of Medical Genetics guidelines for sequence interpretation.
Study population
With the inclusion and exclusion criteria mentioned above, the study population did have some notable differences between cohorts and characteristics that may be specific to CUMC. For example, the CUMC cohort, for which actionable genes could be further interrogated due to different consent protocols, included younger adults than AURORA. The CUMC cohort was more ethnically diverse and also had a disproportionate number of glomerular patients, which is expected due to CUMC’s status as a world-class glomerular disease referral center. These characteristics are marked in red in Table 1.

Table 1 from Groopman et al, NEJM 2019
Study findings
The investigators detected diagnostic variants in 9.3% of their entire cohort, and these variants span 66 different monogenic disorders. Six diseases accounted for 63% of the genetic diagnoses, with mutations in PKD1 affecting the largest number of patients (Figure 1, green star).
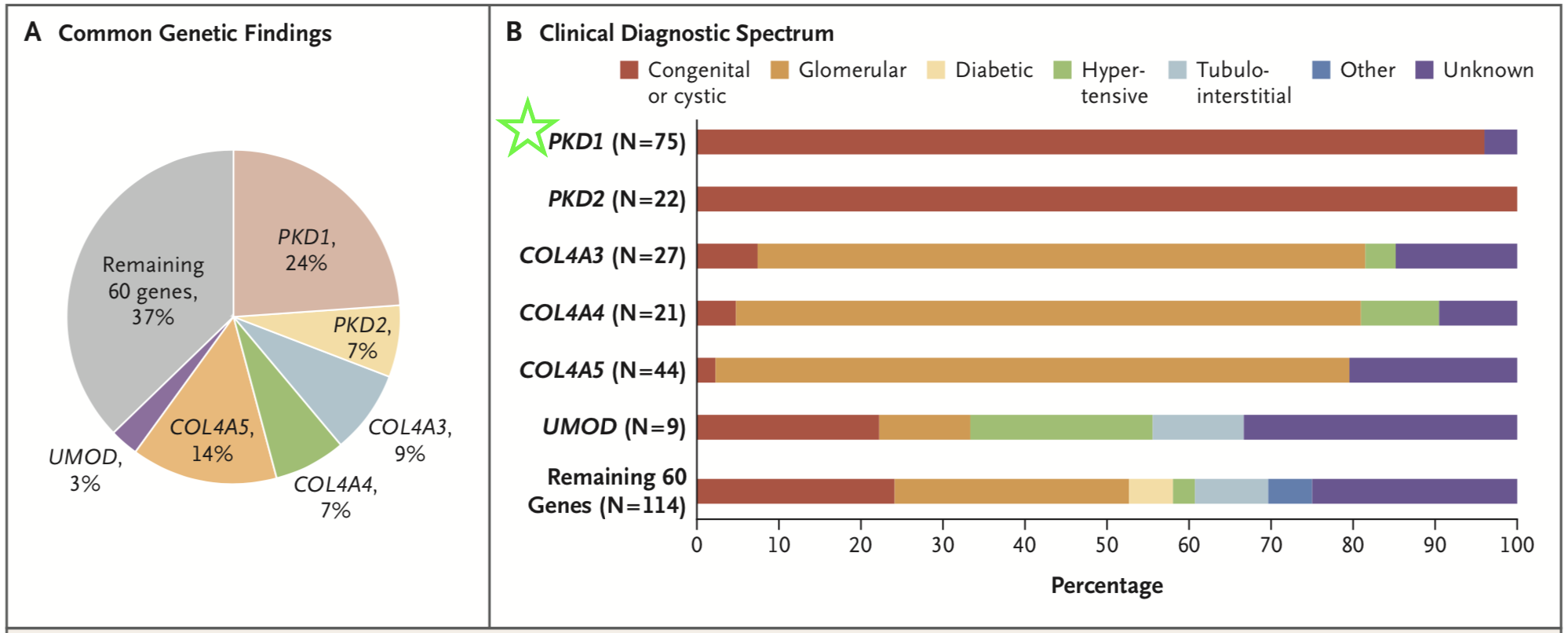
Figure 1 1 from Groopman et al, NEJM 2019
Interestingly, the authors noted that diagnostic yield was highest in patients congenital or cystic renal disease (23.9%) and in patients with “nephropathy of unknown origin” (17.1%) (Table 2, green stars).

Table 2 from Groopman et al, NEJM 2019
Breaking this down further, it appears that the highest diagnostic yield for nephropathy of unknown origin is actually in the AURORA cohort, whereas there was a lower diagnostic yield for glomerulopathy compared to tubulointerstitial disease in the CUMC cohort, which may not be surprising given the local glomerular expertise there. There was still a high predictive diagnostic yield for glomerulopathy in the AURORA cohort, which would be more representative of a community population (Table 3, red box).
To summarize:
62% of patients with COL4A3, COL4A4, or COL4A5 pathogenic variants did not have clinical diagnoses of Alport syndrome or thin basement membrane disease but had the diagnosis of focal segmental glomerulosclerosis (FSGS).
With exome sequencing, 18 CUMC patients were re-classified from FSGS to nephropathy associated with COL4A3, COL4A4, or COL4A5.
For 65 CUMC patients, a subtype of disease was identified, for more precise diagnosis and management.
39 CUMC patients with “nephropathy of unknown origin” had a molecular cause of their CKD identified.
For 89% of the CUMC patients with genetic findings from the exome sequencing, the molecular diagnosis has implications to alter their clinical management (Table 4, red box).

Table 4 from Groopman et al, NEJM 2019
Food for thought
The findings of this study emphasize the high degree of genetic and phenotypic heterogeneity of kidney disease and show the extent to which genetic testing can help resolve clinical diagnostic challenges. However, we have to keep in mind that genetic testing does not always give complete answers but does have the potential to alter medical management when interpreted in the appropriate clinical context. Given the abundance of rare, predicted damaging variants in the human genome, the risk of falsely attributing causality is always high. Physicians and geneticists must incorporate diagnostic sequence interpretation with traditional tools such as clinical history and renal biopsy, as well as with other sources of omics data, all of which can provide crucial insight into the genetic findings. Thus, this multidisciplinary approach improves the diagnostic utility of exome sequencing in different clinical categories of kidney disease, as reported in this study.
For more discussion, please join us for the NephJC chats this week!
Summary drafted by Yasar Caliskan,
WashU at St Louis and NSMC intern, class of 2019
Edited by Jennie Lin, MD MTR,
Attending Nephrologist and Physician-Scientist
Northwestern University, Chicago
Michelle Rheault breaks down the implications of this week’s paper for us