

The fourteenth #NephJC is November 4 at 9 EST
How I stopped avoiding immunology and learned to love the T cell

The Talented Dr. Steigerwalt
I am a Clinical Hypertension Specialist (with a secret identity as a blood and guts nephrologist) There have been rumblings for many years that inflammation and hypertension are intertwined. The mechanism, other than increased reactive oxygen species, was unclear. Along comes Dr. David Harrison’s lab at Vanderbilt, and discovery after discovery uncovers an amazing story, which almost reads like a novel, implicating activated T cells in the pathogenesis of most forms of hypertension- at least in mice and rats-and portends focused therapy to reduce blood pressure without eliminating immune function.
I recommend reading The immune system in hypertension by Trott and Harrison for background of CNS sympathetic activity and T cell activation; below is the background you will need as a non-immunologist to work through the JCI article we are reviewing for journal club.
Definitions: Our immune system is composed of innate (present from birth) and adaptive (acquired) immunity. (Up to date™ has excruciating detail if you are interested.)
T and B lymphocyte cells (produced in the bone marrow) make up our adaptive immune system. Each T and B lymphocyte acquires a structurally unique receptor during development. From this vast repertoire cells exposed to antigens multiply and expand a specific group of identical cells (clone)
“Each naïve T cell expresses a unique TCR (T-cell receptor) which is specific for a small number of closely related short peptides in a complex with a polymorphic membrane proteins encoded by genes within the MHC (major histocompatibility complex). Some T cells recognize modified self peptides; i.e. a class ll MHC molecule, HLA-DR-4, is strongly associated with RA. A link to specific MHC alleles is a common feature of autoimmunity.”
Naïve T cells must receive 3 signals in order to mobilize:
- Specific antigen (the peptide bound to the major histocompatibility complex (MHC))
- Antigen independent boosters or co-stimulators (CD80 or CD86) and
- Cytokines that promote cell expansion and differentiation
There are 3 types of antigen presenting cells: macrophages, B cells and dendritic cells.
DC (dendritic cells) are the major antigen presenting cells; they capture, process and present antigens to T cells in order to induce adaptive immunity against a specific antigen. DC activating signals include PAMPS (pathogen associated molecular panels) and DAMPS (damage associated molecular panels). PAMPs and DAMPs localize DC to the site of infection or injury, respectively. DCs acquire the antigen before migrating to secondary lymphoid organs such as spleen or lymph nodes.
At the secondary lymphoid organ, the antigen is presented to naïve T cells with a relevant TCR (T cell receptor). Antigen activated T cells proliferate, inducing a clone of daughter cells specific for the antigen which differentiate into effector cells, producing cytokines. A few become long-lived memory cells.
Major Histocompatibility complex- is an area of the genome, which codes for a series of proteins expressed on cells. These cells serve as flags to distinguish between self and non-self (foreign) proteins.
Previous work by Harrison’s lab has shown that hypertension in mice caused by DOCA -salt (analogous to hyperaldosteronism) and also Angiotensin-ll infusion requires the presence of T cells. Mice lacking T cells do not become hypertensive in response to these stimuli.
This week's JCI article reviews a series of experiments further defining the role of the T cell and identifying the modified self antigen that initiates the autoimmune response resulting in hypertension in laboratory animals and also includes human data identifying the antigen in humans with hypertension. Walk in the Park.

DC isoketal-modified proteins activate T cells and promote hypertension
Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd, Harrison DG.
PMID: 25244096
Previous work/introduction:
T cell macrophages accumulate in blood vessels and kidneys of experimental animals with hypertension.
Immunosuppression ameliorates hypertension in experimental animals and in humans with RA and psoriatic arthritis.
Mice lacking lymphocytes (Rag -/-) are protected from vascular dysfunction and oxidative stress in response to Angiotensin-ll, Norepi and DOCA–salt. Transfer of T cells but not B cells restores the hypertensive effect of all three. Blockade of co-stimulatory molecules CD80 and CD86 also prevents hypertension and T cell activation.
ROS (reactive oxygen species) produced by NADPH oxidases are involved in hypertension and end organ damage. Mice lacking components of NADPH oxidases are protected against hypertension and target organ damage.
Isoketals are formed by free radical lipid peroxidation. Increased production of isoketals has previously been found in diseases with oxidative stress, including alcoholic liver disease, atherosclerosis, Alzheimer’s .
This article shows that hypertension causes isoketals to accumulate in DCs and promote DC cytokine production and immunogenicity. Compounds that scavenge isoketals prevent DC activation and experimental hypertension.
Results: A logical series of experiments :
Experiment 1:

Figure 1.
See figure 1: key points: in WT (wild type) mice vs. Nox2–/– ( mice lacking a component of NADPH oxidase and unable to develop oxidative stress) the WT increase superoxide. Isoketal staining heart and aorta was present in WT animals. A scavenger of isoketals, 2-HOBA, prevented accumulation of isoketals in heart and aorta.
Experiment 2:
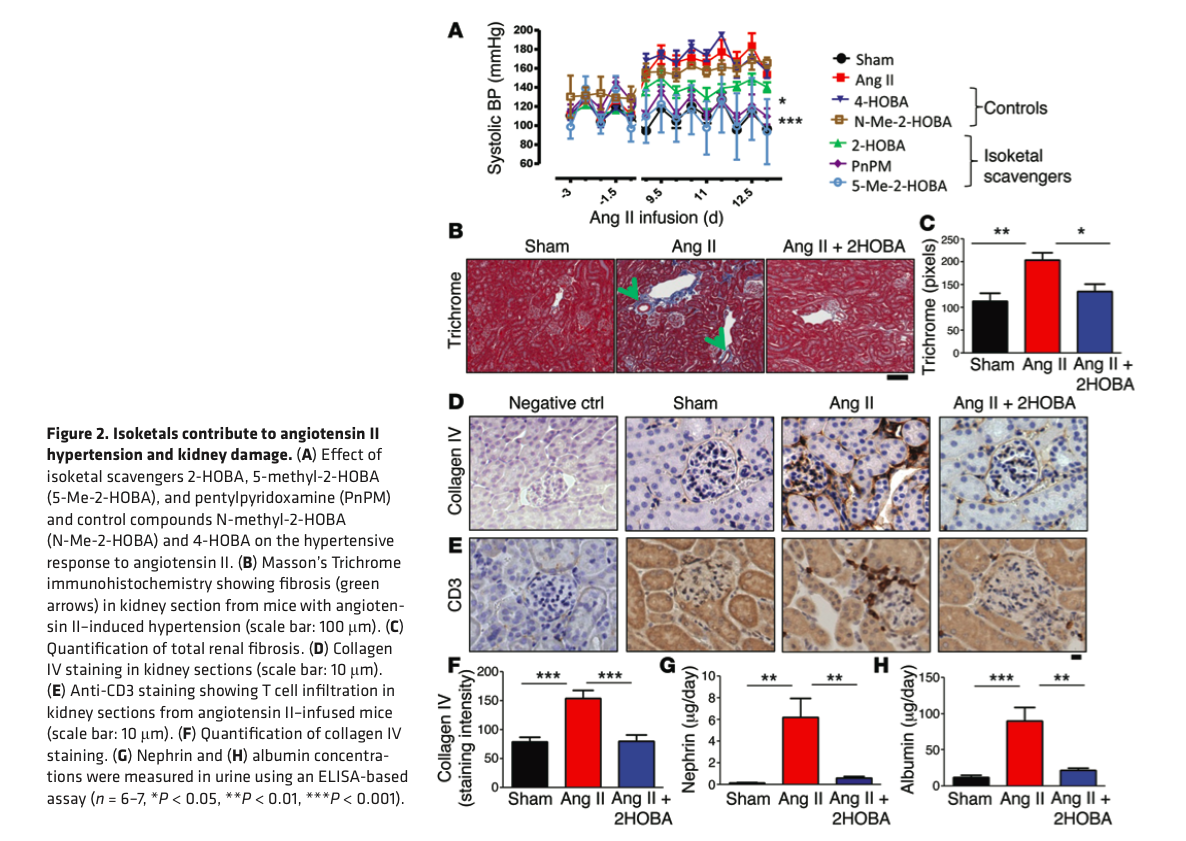
Figure 2
See figure 2: Isoketals contribute to angiotensin ll mediated renal fibrosis (accumulation in the kidney of type 4 collagen), albuminuria and hypertension: this is blocked by the isoketal scavenger 2-HOBA.
Experiment 3

Figure 4
See figure 4: Isoketals accumulate and activate DCs in hypertension: they used flow cytometry using a gating strategy to look at cells with CD11b and CD11c (which are present on DCs). CD80 + DCs also accumulate isoketals in Angiotensin ll induced HTN.
Experiment 4
Maturation of DCs is associated with increase surface expression of CD80 and 86. Cells staining positive for isoketal protein adducts increased striking increase in surface expression of CD80 and CD86; most prominenetly on CD11b+ /CD11c+DCs and was prevented buy 2-HOBA ( figure 4 )
Experiment 5
Measured cytokine production in DCs from ang ll infused mice; markedly increased including Il-6; blocked by 2-HOBA.
Experiment 6

Figure 5.
Repeated experiment 5 in DOCA-salt mice. Increased production of isoketals was found, associated with HTN, which was decreased by 2- HOBA (isoketal scavenger); isoketal accumulation also blocked by 2-HOBA. See Figure 5.
Experiment 7

Figure 6.
Tried to determine whether DCs from Ang ll treated mice can activate t cells from untreated mice. DCs from these mice caused proliferation and survical of CD8 + t cells. Figure 6
Experiment 8
Expression of five hundred five genes was altered in mice made hypertensive for 2 weeks by Angiotensin ll infusion. 150 were not altered in Nox2 –/– mice (remember, they can not produce reactive oxygen species, ROS) and 141 normalized by 2-HOBA treatment. See supplemental table 2. Thus many, but not all, gene changes associated with angiotensin- ll induced HTN were associated with ROS and isoketal production.
Experiment 9
Was performed to determine HOW isoketals alter DC immunogenicity. To determine whether isoketals could act as neoantigens, mouse kidney homogenates were exposed to isoketals, from ang ll hypertensive mice; and 3 other similar proteins were used as controls. (see figure 6) .The experiment showed that DCs from Ang ll mice promote T cell survival, proliferation and cytokine production.
Experiment 10
Surface expression of MHC1 was examined; increased expression of 2 proteins: H-2Kb and H-2Db were found (done via measurement of florescence; supplemental figure 5a) among CD11c cells (DC cells) from Ang ll mice.
Treatment with 2-HOBA prevented these changes. Thus MHC1 surface repertoire is altered by hypertension.
Experiment 10
Because DCs from hypertensive mice seemed to present isoketal modified proteins more efficiently, similar peptides were tested but DCs exposed to these proteins did not show this increased efficiency. This shows that this is a unique property of isoketals.
Experiment 11

Figure 9.
Transfer of hypertension by DCs: DCs from hypertensive Ang ll mice when transferred to non hypertensive mice resulted in enhanced hypertensive effect of Ang ll, whereas DCs from sham treated mice transferred to non hypertensive mice did not show a similar increase in sensitivity of mice to ang ll. The Ang ll hypersensitivity was blocked by co-incubation with 2-HOBA. See Figure 9.
Additional experiments were performed on Rag-/- mice (no lymphocytes) and despite DCs from Ang ll treated mice BP sensitivity was not enhanced proving the need for T cells to mediate the changes.
Experiment 12
Induction of oxidative stress in DCs promotes isoketal formation, activation of T cells and hypertension. DCs ex vivo were treated by oxidants which resulted in formation of intracellular isoketals, increased surface expression of CD86 (figure 9). Co-culture of these cells with T cells promoted survival and proliferation of CD8+ T cells. Transfer of the activated DC cells increased BP response to low dose Ang ll in recipient mice as in previous experiment These changes were prevented by co-administration of 2-HOBA.
Human data
Increased F2 isoprostanes (these are compounds formed in vivo from free radical catalyzed peroxidation of essential fatty acids - isoketals are related compounds) in hypertensive humans: do isoketals play a role in human hypertension?
In order to determine this, these were measured in 3 cohorts of patients: normotensive, controlled hypertension (2-3 meds) and resistant hypertension (3-4 meds)-see table 2. F2 isoprostanes were significantly elevated in resistant hypertension compared to normotensive and controlled hypertensives. (table 3)


A separate experiment using buffy coats in 8 normotensive and 12 controlled hypertensives showed presence of isoketal adducts in hypertensive were three-times greater than in normotensives.
Discussion:
This article shows a new pathway for hypertension from the cellular level to the whole animal to the human with a range of nuanced experiments weaving a beautiful story. Make sure you don't miss the editorial and supplemental readings so it makes sense. And perhaps we will soon target T8+ lymphocytes in patients with resistant hypertension.