
#NephJC Chat
Tuesday July 9, 2019 at 9 pm Eastern
Wednesday July 10, 2019 at 9 pm Indian Standard Time
Wednesday July 10, 2019 at 9 pm British Summer Time, 1 pm Pacific
N Engl J Med. 2019 Jul 4;381(1):36-46. doi: 10.1056/NEJMoa1814427.
Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy
Fervenza FC, Appel GB, Barbour SJ, Rovin BH, Lafayette RA, Aslam N, Jefferson JA, Gipson PE, Rizk DV, Sedor JR, Simon JF, McCarthy ET, Brenchley P, Sethi S, Avila-Casado C, Beanlands H, Lieske JC, Philibert D, Li T, Thomas LF, Green DF, Juncos LA, Beara-Lasic L, Blumenthal SS, Sussman AN, Erickson SB, Hladunewich M, Canetta PA, Hebert LA, Leung N, Radhakrishnan J, Reich HN, Parikh SV, Gipson DS, Lee DK, da Costa BR, Jüni P, Cattran DC; MENTOR Investigators.
PMID: 31269364 Full Text at NEJM


Introduction
2019 has been an exciting year for patient-oriented kidney research, and the excitement continues with the highly anticipated Membranous Nephropathy Trial of Rituximab (MENTOR) study, published in the NEJM. This study adds to the growing momentum of B-cell biology that has revolutionized our understanding of primary membranous nephropathy (PMN) and shifted our approach to studying and treating PMN.
Primary membranous nephropathy (PMN) is a kidney-specific, autoimmune disease resulting from subepithelial deposition of IgG and complement components along the glomerular basement membrane. Below we can see examples of patient biopsies: granular capillary loop immunofluorescence (IF) on the left and electron microscopy (EM) showing subepithelial deposits on the right.

Here is another example of IF of PMN.

Courtesy of Arkana Labs. Granular capillary loop IF seen in PMN.
PMN is one of the leading causes of non-diabetic nephrotic syndrome among adults, with an incidence of 12 to 20 per million per year in the United States. It occurs in multiple ethnicities but is most commonly seen in white patients in the fifth and sixth decades of life, with a 2:1 male predominance. Patients typically present with nephrotic-range proteinuria and its associated symptoms of edema, hypoalbuminemia, and hyperlipidemia, although a subset of patients have stable subnephrotic proteinuria and never progress to end-stage kidney disease (ESKD). Discussion of secondary membranous nephropathy is outside the scope of this summary but can be reviewed here and here.
Due to variable PMN disease course and toxic side effects of traditional immunosuppressive therapy, the current consensus Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis (GN) approach to treating PMN is to first initiate conservative supportive therapy with angiotensin-converting-enzyme (ACE) inhibitors or angiotensin-receptor blockers (ARBs) and by controlling edema and hyperlipidemia. Use of immunosuppressive therapy is reserved for patients with persistent nephrotic-range proteinuria. Because glucocorticoids as monotherapy does not work (see Missteps in Nephrology region for NephMadness 2016), immunosuppressive therapy for PMN has traditionally involved the Ponticelli protocol of cyclical alkylating agents (cyclophosphamide or chlorambucil) accompanied by pulses of methylprednisolone (summarized here). Calcineurin inhibitors such as cyclosporine and tacrolimus can also induce remission but the remission tends to be temporary with high rates of disease recurrence. Additionally calcineurin inhibitors have nephrotoxic side effects. In this context, the Membranous Nephropathy Trial of Rituximab (MENTOR) study investigates whether B-cell depletion with rituximab, which has a more tolerable side effect profile than alkylating agents, is noninferior to calcineurin inhibition with cyclosporine in inducing and maintaining remission of PMN.
To understand the rationale for and significance of MENTOR, one must first understand the history behind our understanding of PMN pathophysiology.
In 1957, pathologist David Jones first identified membranous GN as a separate entity from other lesions associated with nephrotic syndrome. Using a special periodic acid–silver methenamine stain, now known as the Jones stain, Dr. Jones reported the unique features of MN not shared by what is now known as membranoproliferative GN, minimal change disease, and focal segmental glomerulosclerosis (FSGS). In his report, he wrote: “In the present investigation it has become apparent that this lesion is the result of a peculiar modification of the epithelial basement membrane of the glomerulus. With the periodic acid silver methenamine stain this thickened membrane resolves itself into a much less thick epithelial membrane from which protrudes externally innumerable short silver-positive projections of club or mushroom shape. Between these projections is a silver-negative hyaline material which is in droplet form (Fig. 10).”

Here is another example of silver staining.

Courtesy of Arkana Labs.
In the 1957 manuscript, Dr. Jones also states, “An auto-antibody formation similar to that of acute glomerulonephritis appears to be present but its mechanism is still unknown.” This concept of autoimmunity in nephrosis extended into experimental science in 1959 with the development of a rat experimental model subsequently known as Heymann nephritis, where intraperitoneal injection of rat proximal tubular homogenates into other rats induced clinical and histopathological features similar to human MN. In other words, injecting rats into their peritoneal space with ground up proximal tubules of other rats induced an inflammatory reaction leading to subepithelial deposits akin to those seen in human MN. Because the antigen(s) from the ground up kidneys did not cross the peritoneal space, those immune complexes in the subepithelial deposits formed from an auto-antigen. Until the 1970s, nephrologists believed these autoimmune complexes were circulating immune complexes passively trapped by glomeruli. Then, in the late 1970s additional seminal studies (including one from Dr. David Salant) established that subepithelial deposits can be induced by in situ immune complex formation rather than circulating immune complex deposition. In other words, the subepithelial deposits seen in MN could be induced by antibodies binding to an antigen residing on the glomerular capillary wall, potentially originating from the podocyte. This concept is best illustrated by the figure below, where panel A is a representation of circulating immune complex deposition while panel B is a representation of how in situ immune complex formation works, with the antigen originating from the visceral epithelial podocyte.

From Glassock RJ, NEJM, 2009.
During the 1980s, scientists attempted to identify this podocyte-intrinsic antigen, and from the Heymann nephritis model they believed it was megalin, a member of the low-density lipoprotein (LDL) receptor family, that in rats is expressed on both podocyte foot processes and the proximal tubule. However, in humans, podocytes do not express megalin, and a search for anti-megalin antibodies in PMN patients turned up empty. Enthusiasm for the paradigm-shifting in situ immune complex hypothesis began to wane until a 2002 report in the NEJM attributed a case of neonatal MN to transplacental passage of maternal antibodies against neutrophil endopeptidase on the baby’s podocytes.
Several years later (50 years after the first description of the Heymann nephritis model), Drs. Larry Beck (@LaurenceHBeckJ1) and David Salant reported in a landmark NEJM study that the M-Type phospholipase A2 receptor (PLA2R) is a major target antigen in human PMN. Years of experimentation exposing extracts of healthy human glomeruli to PMN patient sera led to this discovery through a combination of Western blotting (to assess whether PMN sera binds to any glomerular proteins), mass spectrometry (to identify what this bound glomerular protein is), and immunoprecipitation with anti-PLA2R antibody (to validate the target antigen). They found that the majority of sera from sampled PMN patients reacted with PLA2R but that sera from patients with FSGS, diabetic nephropathy, or no kidney disease did not. Three decades after the initial in situ immune complex hypothesis for PMN emerged, a specific causal antigen and causal biomarker (circulating antibody) were finally identified. The excitement in the nephrology world was palpable, as evident in the late Dr. Nate Hellman’s Renal Fellow Network posts on the initial unveiling of unpublished results and on the NEJM report almost exactly 10 years ago.

Courtesy of Arkana Labs. Strong anti-PLA2R staining on IF of a PMN biopsy.
In addition, the presence of anti-PLA2R in the serum of a PMN patient correlated with disease activity, as shown in Figure 6 of the 2009 report below. The red box in panel B highlights the presence of anti-PLA2R activity (2 dark bands in the box) in December 2005, prior to the patient’s initiation of immunosuppressive treatment (charted in panel A). Of note, the antibody disappears by April 2006, prior to full remission of proteinuria. We will return to this detail later.

Salant’s group also identified another putative antigen in a small number of anti-PLA2R negative cases that ended up being thrombospondin type 1 domain-containing 7A (THSD7A).

Courtesy of Arkana Labs. Strong anti-THSD7A staining on IF of a PMN biopsy.
So what does this scientific history lesson tell us? Decades of research have informed us that PMN is an autoimmune disease with podocyte-intrinsic antigens that form in situ subepithelial immune complexes with auto-antibodies against these antigens. We now have disease-specific, causal biomarkers of PMN, meaning that we have biomarkers (anti-PLA2R and anti-THSD7A) that directly contribute to disease pathogenesis and are not found in other glomerular diseases. Because the antibody is causal, why not treat PMN by eliminating production of the disease-causing antibody? Because the antibody is causal, why not track disease status by circulating antibody levels?
Let’s start with the first question. Yes, it would make sense that immunosuppressive therapy works by decreasing production of the disease-causing antibody, but not all immunosuppressive therapies are equal. Nephrologists have been investigating the role of rituximab in treating PMN because rituximab targets CD20 on B cells and depletes B-cell lineages that would later mature into antibody-producing cells as illustrated in the adapted figure below.
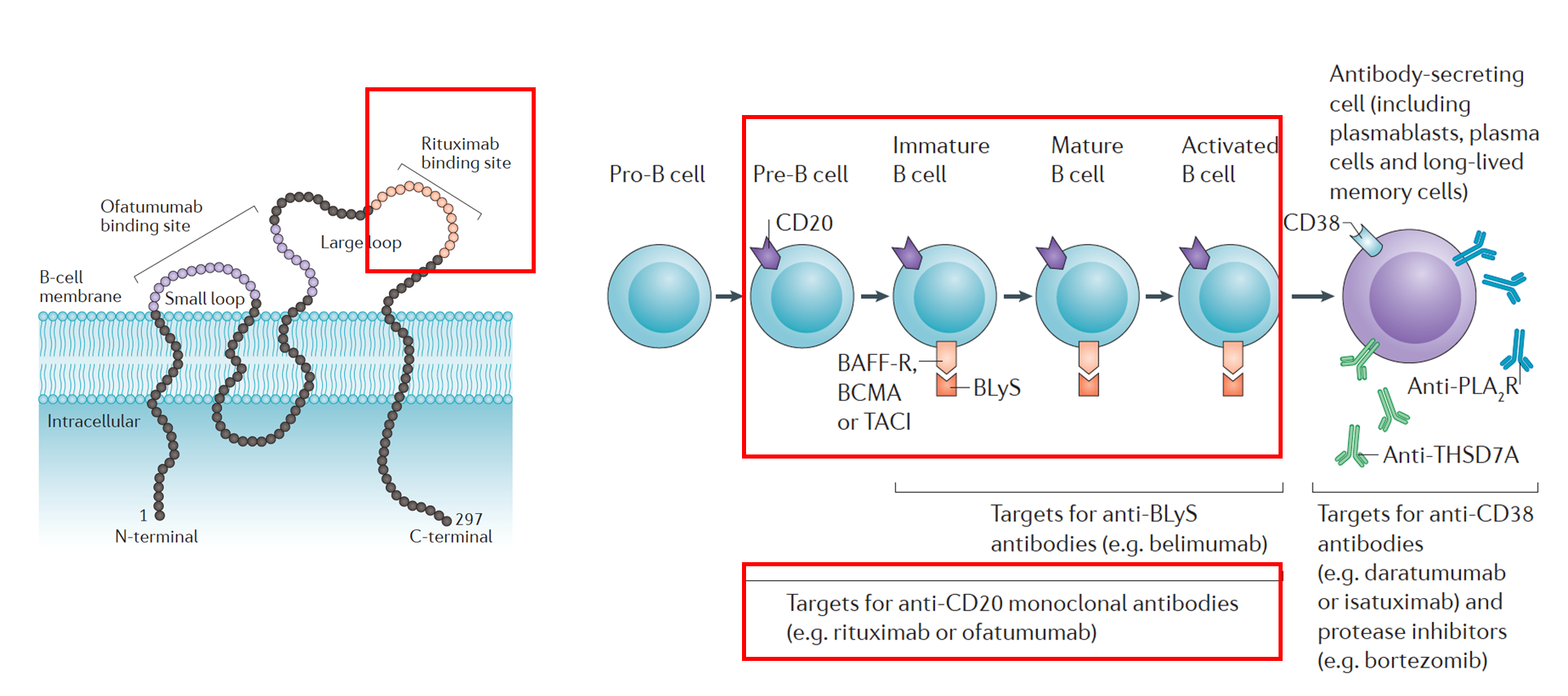
Adapted from Ruggenenti et al., Nature Reviews Nephrology, 2017. The left panel shows the rituximab binding site on the large loop of the CD20 molecule of a B cell. The right panel shows which types of cells would be targeted by rituximab: CD20 positive pre-B cells, immature B cells, mature B cells, and activated B cells. Plasma cells and long-lived memory cells would not be targeted due to absence of CD20.
With a more specific mechanism of action than the current first-line alkylating agents, rituximab also has a more mild side effect profile, making it a more attractive therapeutic option should RCT data support its efficacy. Prior to the MENTOR study, clinical trials investigating the effect on rituximab and PMN remission have been small but promising. In the GEMRITUX study that initially enrolled 80 patients, rituximab failed to meet the primary end point of complete or partial remission of nephrotic syndrome at 6 months, although remission was achieved in the post randomized control trial (RCT) period. The study did find that compared to conservative therapy alone, rituximab significantly decreased circulating anti-PLA2R antibody levels within 3 months. In retrospect, this lag time between depletion of circulating anti-PLA2R antibody and remission of proteinuria would have been expected based on Beck’s initial 2009 NEJM report, which presented a more rapid decline in serum antibodies prior to substantial decrease in proteinuria (see brief summary of that report above). Indeed, this phenomenon was confirmed in a subsequent report. For the “negative” GEMRITUX study, was 6 months a reasonable primary outcome time point for remission of proteinuria, and are anti-PLA2R levels a more optimal functional biomarker for trial outcomes? People were still willing to bet on rituximab, and the trial provided a lesson in restrategizing how to run an RCT in PMN.
As mentioned above, alkylating agents such as cyclophosphamide have historically been part of the gold standard for treating PMN patients with persistent nephrotic-range proteinuria not responsive to conservative therapy. However, due to its blunt mechanism of action, which involves inducing DNA damage in all proliferating cells and inhibiting B cell function, cyclophosphamide has an unfavorable side effect profile that gives many physicians pause before prescribing it for PMN. Rituximab, with its more specific mechanism of action targeting B cells rather than all proliferating cells, has a less severe side effect profile. Indeed, the two agents were compared in a prospective observational cohort study in terms of safety profile and remission rates, and patients receiving rituximab were found to have fewer adverse events while experiencing similar complete disease remission rates. At the moment, alkylating agents, not rituximab, are still indicated by KDIGO as first-line treatment for the subset of PMN patients with persistent nephrotic range proteinuria as we await more RCT data on rituximab, but could the MENTOR study help change that?
Meanwhile, calcineurin inhibitors (CNIs), which have significant although less severe side effects than cyclophosphamide, have become an increasingly popular therapeutic option for PMN among nephrologists in the United States and elsewhere. Cyclosporine and tacrolimus have comparable efficacy when compared against each other. Like cyclophosphamide, their main mechanism of action (reviewed here and here) is not B-cell specific, but compared to cyclophosphamide, CNIs have higher rates of relapse after drug withdrawal.
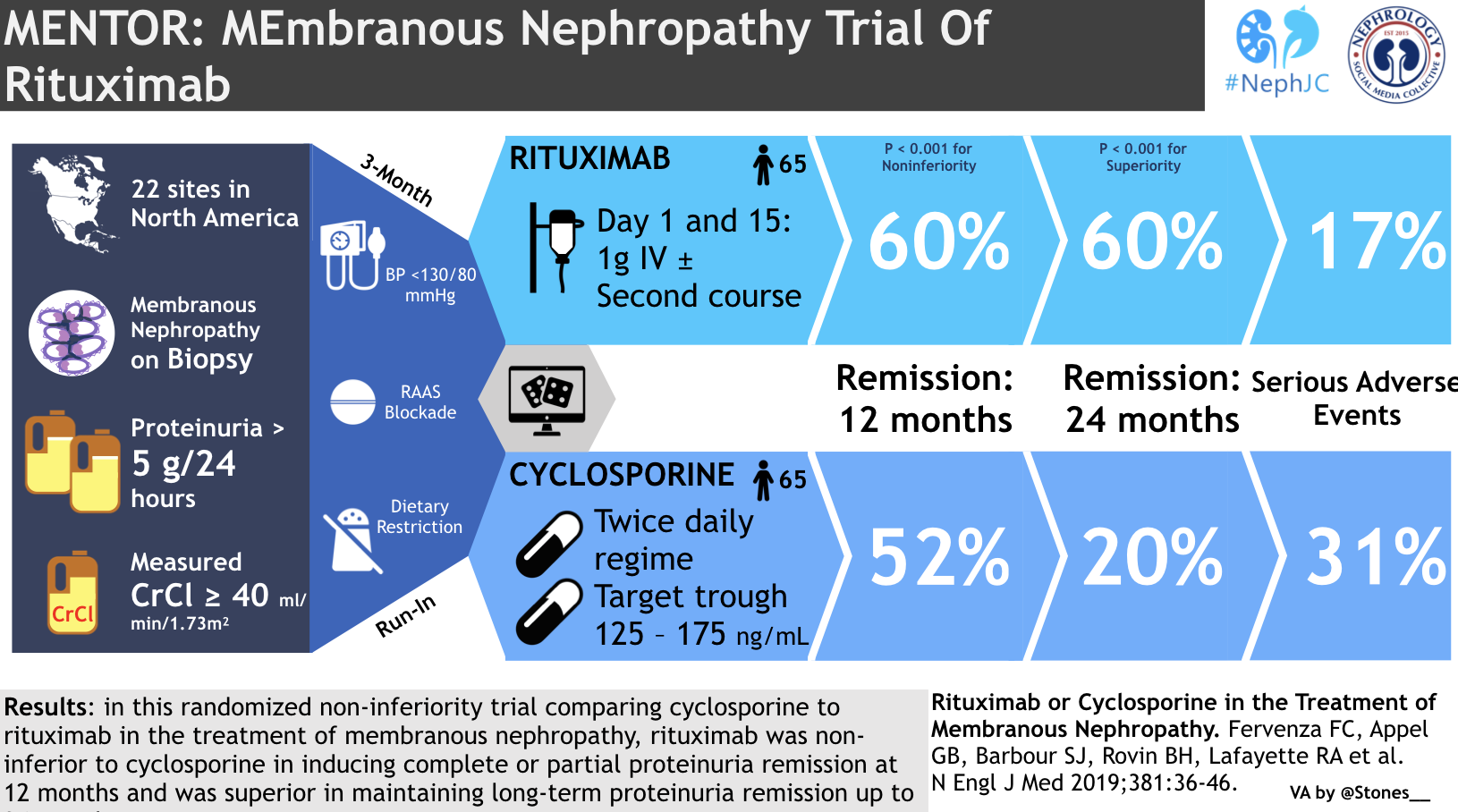
So now 60 years after the first description of Heymann nephritis and 10 years after the published discovery of PLA2R, we have the MENTOR study, which compares CNI cyclosporine with the B-cell depletion strategy of rituximab in the treatment of PMN. The MENTOR investigators set out to ask whether rituximab is noninferior to cyclosporine in inducing and maintaining remission of proteinuria for up to 24 months in patients with PMN. Below we will discuss the study design, results, and implications for future PMN studies and management.
For more background on PMN, we recommend checking out this CJASN review by Couser; CJASN review (more cautious about rituximab) by Ponticelli and @GlassockJ; AJKD retrospective by @GlassockJ; AJKD review by Francis, Beck, and Salant; RFN post by @Slatts_1 ; most recent KDIGO statement, and this GlomCon lecture.
The Study
The MENTOR study set out to answer the following question: Is rituximab therapy noninferior to cyclosporine therapy in inducing and maintaining remission of proteinuria, regardless of baseline anti-PLA2R status, for up to 24 months in patients with PMN?
Trial Design
To answer this question, the investigators designed an investigator-initiated, open-label, multicenter, randomized noninferiority trial. The purpose of a noninferiority trial is to evaluate whether a new treatment (rituximab) has similar efficacy to that of an established treatment (cyclosporine, per KDIGO GN guidelines). Noninferiority trials (reviewed here) have become prevalent because treatment with a placebo or no-treatment arm, especially for a disease such as PMN, is not ethical when an effective treatment already exists. For MENTOR, the null hypothesis is that in PMN, rituximab induces and maintains remission of proteinuria at a lower rate than cyclosporine does. Rejection of this null hypothesis by a noninferiority margin of 15% on an absolute risk-difference scale would support that rituximab is noninferior to cyclosporine in the treatment of PMN for the predetermined primary outcome. All participants were either already on conservative therapy (ACEI or ARB) or were placed on conservative therapy during the run-in phase prior to being randomized to either the rituximab arm or the cyclosporine arm. \
Funding Source
Genentech (maker of rituximab) supported the study, although the authors state the company did not influence the planning, data collection, interpretation, or writing of the study.

Figure S1: Overview of Trial Design. NR = non-response, CR = complete remission
Study Participants
Broadly speaking, the participants were adults with biopsy-proven PMN already on conservative management but not on other immunosuppressive agents. The inclusion and exclusion criteria are stated in detail in the supplemental section of the manuscript.
Inclusion Criteria:
PMN diagnosed by kidney biopsy demonstrating subepithelial spikes along capillary walls by silver stain, granular IgG and C3 along capillary walls on IF, and subepithelial deposits seen on EM. All three components were required, and pathology was adjudicated by a study PI prior to randomization.
Age: 18 to 80 years
If female, not pregnant; if not post-menopausal or surgically sterile, must be practicing medically approved contraceptive method
Not exposed to prednisone or mycophenolate mofetil for > 1 month, not exposed to alkylating agents for > 6 months
Treatment with RAS blockade and/or ARB for ≥ 3 months prior to randomization, BP < 140/80 in > 75% readings.
Participants with more than 5g per 24-hours were able to enter the study’s treatment phase without the run-in period
Estimated GFR (by MDRD) ≥ 40 mL/min/1.73m2 or quantified creatinine clearance ≥ 40 mL/min
Exclusion Criteria:
Active infection
Secondary cause of MN (hepatitis B, SLE, medications, malignancy)
Type 1 or type 2 diabetes mellitus to exclude proteinuria due to diabetic nephropathy
Pregnancy or breast feeding
History of resistance to CNIs, rituximab, or alkylating agents (although relapse after 3 months for CNI and 6 months for rituximab and alkylating agents was still accepted for eligibility)
Intervention:
Eligible participants were randomized to either the rituximab arm or the cyclosporine arm and observed for 6 months.
Rituximab dosing: 1000 mg intravenous (donated by Genentech) on days 1 and 15
Cyclosporine dosing: 3.5 mg per kg per day every 12 hours (purchased from Novartis), with target serum trough levels 125 to 175 ng/mL as assessed every 2 weeks until target trough obtained. Dose reduction was performed for participants with a persistent and unexplained increase in serum creatinine of more than 30% (if creatinine did not fall in response to this dose decrease the participant was considered to have experienced treatment failure).
If at 6 months into the study complete remission was observed in either arm, treatment was stopped. In the cyclosporine arm, the drug was then tapered over an additional 2-month period. For both arms, if non-response was observed (reduction in proteinuria by less than 25%), treatment was stopped, and in the cyclosporine arm a 2-month taper was initiated.
At the 6-month mark, partial responders continued for an additional 6 months, with treatment then stopping at the 12-month mark (with relevant taper for cyclosporine). Note how this additional step for months 6-12 differs from the absolute stop at the 6-month mark of the GEMRITUX trial (discussed in our introduction). Additional follow up in MENTOR was performed until month 24.
Sample Size and Stats
130 participants were enrolled and randomized, with n = 65 in each arm. The investigators calculated that they would need 63 participants in each arm to achieve 80% power to detect noninferiority at a one-sided alpha of 0.025 corresponding to a two-sided alpha of 0.05 and a noninferiority margin of 15% on an absolute risk difference scale. In other words, this study was powered for the desired, preset outcome. As a side note, one major limitation of approximately half of noninferiority trial is the failure to state the a priori noninferiority margin, so the explicit statement of the proposed margin in the main text and supplement is already a strength. However, the best practice for defining such a margin is less clear. It is generally recommended that the margin “be defined based on statistical considerations and clinical judgement” where a margin can be defined based on a pooled estimate of effect size from historical placebo-controlled RCTs or based on the limit of the confidence interval (CI) closest to the null effect. For the MENTOR study, the 15% corresponded to half of the risk difference of approximately 30% expected at month 24 compared with placebo, according to a prior cyclosporine RCT. The margin was also determined by the rationale that benefits of rituximab--tolerability, less nephrotoxicity, greater compliance, and lower cumulative cost of discrete injections over daily medications--would justify a 15% inferiority margin for efficacy. To run the statistics on whether rituximab was noninferior, standard error for Z tests comparing differences between groups was calculated using a generalized linear model with binomial distribution and identity link function. The test was considered significant if the one-sided p value was < 0.025. If noninferiority was established, then superiority was tested. Rituximab was considered superior to rituximab if a two-sided p value was < 0.05. Other comparisons (Kaplan Meier curves and hazard ratios from Cox regression models) were performed using 95% CI. Sensitivity analyses were also executed to see whether different statistical approaches would yield the same result.
Outcomes
The primary endpoint was assessed in an intention to treat analysis and was defined as a composite of complete remission or partial remission of proteinuria at 24 months after randomization.
Complete remission was defined as ≤ 0.3 g/24 hours proteinuria and serum albumin ≥ 3.5 g/dL. Partial remission was defined as a reduction in baseline proteinuria of ≥ 50% and final proteinuria between 0.3 and 3.5 g/24 hours (regardless of serum albumin). Neither of these endpoints were defined by eGFR.
Treatment failures consisted of any of the following:
< 25% reduction of baseline proteinuria at 6 months
Relapse
Premature termination before 12 months due to disease activity or adverse event
Patients requiring additional immunosuppressive medications not on the study protocol
Not meeting criteria of complete or partial remission by 24 months
Loss to follow-up
The investigators also examined many secondary outcomes, including:
Different time points for proteinuria remission
Time to remission
Relapse
Treatment failure at different time points
Time to treatment failure
Anti-PLA2R levels at various time points (see potential rationale here)
Quality of life as assessed by the Kidney Disease Quality of Life Short Form (KDQOL-SF)
Decrease in creatinine clearance
ESKD at 24 months
Adverse events
Results
As shown below in Figure S2 of the study, a total of 182 patients were screened, and 130 were enrolled (n=65 in each arm). Out of the 52 excluded, the vast majority did not meet inclusion criteria. After randomization, the rituximab arm had incomplete follow-up on only two participants (considered treatment failure), and in the cyclosporine arm only four participants had incomplete data (considered treatment failure). No participants were excluded from the final analysis.

Figure S2. Overview of recruitment and follow-up.
Participants in each study arm were fairly evenly matched in terms of baseline characteristics, listed in Table 1. They were primarily middle-aged, majority male, with heavy proteinuria (median 8.9 g/24 hours in each arm) and relatively high creatinine clearance (> 80 mL/min in each arm). There was no mention of race or ethnicity, although given the epidemiology of PMN the cohort participants were most likely of primarily European ancestry.
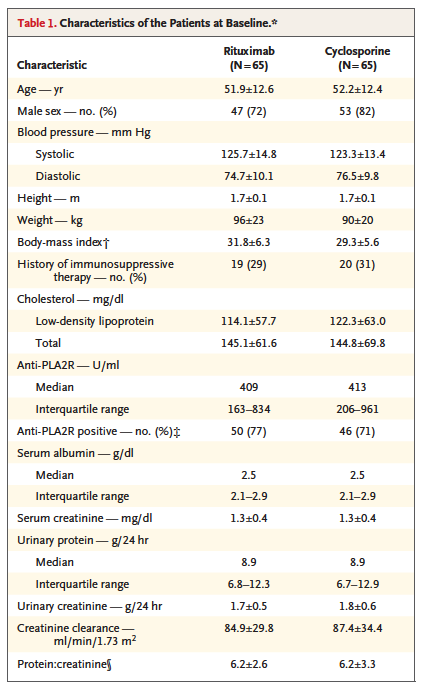
When baseline characteristics were further broken down by sex and randomization arm, median baseline anti-PLA2R levels were 179 u/mL among females in the rituximab arm, compared to 382 u/mL among males in the same arm and 535 u/mL among females in the cyclosporine arm.

So how did rituximab fare against cyclosporine in this study? At the end of 12 months, rituximab was noninferior to cyclosporine in terms of the composite primary outcome of complete or partial remission of proteinuria (60% achieved the composite outcome in the rituximab arm vs. 52% for cyclosporine, P = 0.004). Figure 1 below shows the range (blue line) of risk difference never crossed the dashed noninferiority margin line. Remarkably and perhaps more excitingly, at 24 months, rituximab was superior to cyclosporine in maintaining the composite outcome, remaining at 60% vs cyclosporine’s drop down to 20% with a risk difference (blue line) of 40% (P < 0.001 for both superiority and noninferiority).

Figure 1. The primary outcome: composite of complete or partial remission of proteinuria at 12 and 24 months.
Similar results were seen at month 18 as well (Table 2), and in terms of complete remission at 24 months none in the cyclosporine group had it whereas 35% did in the rituximab group (Table S7). For participants meeting the composite outcome, proteinuria levels (median and interquartile range) were plotted on a log scale in Figure S8 according to treatment arm and time.

The rituximab advantage was consistent across subgroups, although an interaction for sex in the rituximab group disappeared after adjustment for baseline anti-PLA2R (see Table S5 above for different baselines, Figure S5 below for the subgroup data).
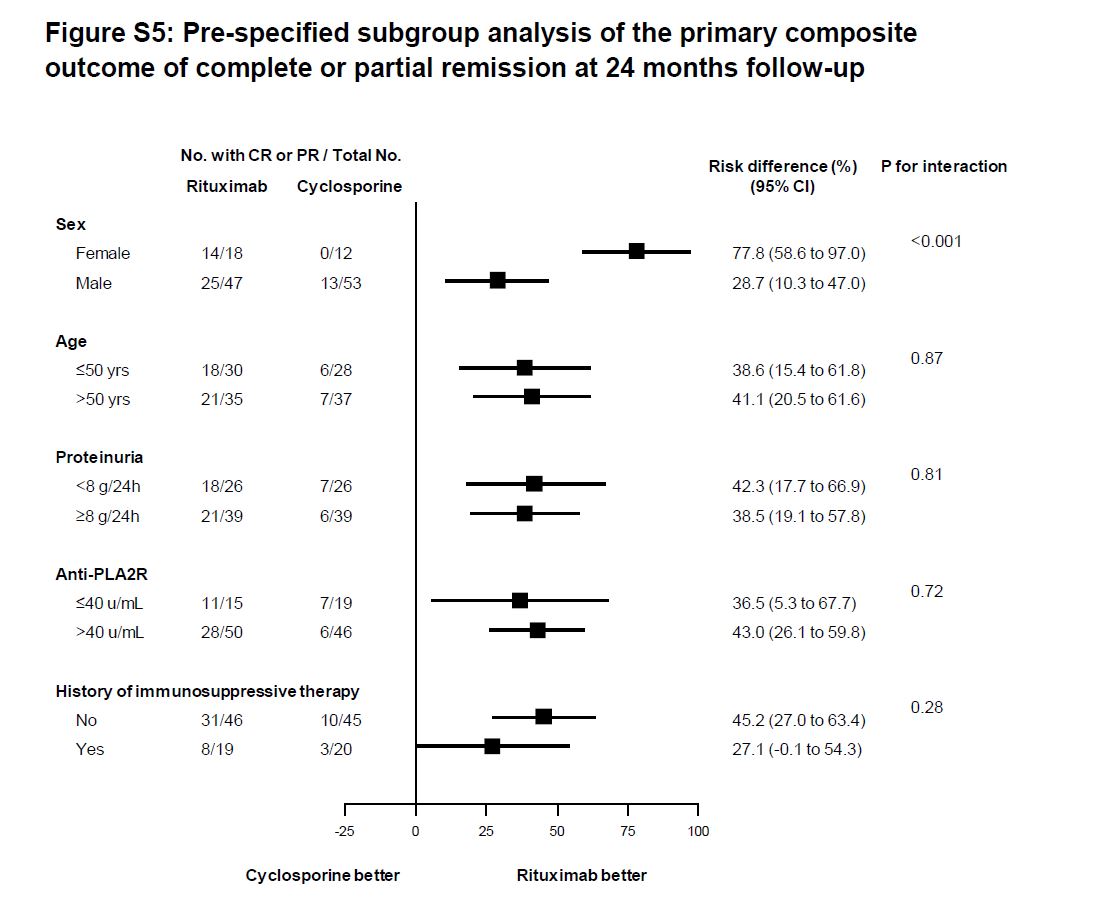
As a secondary outcome of measuring disease activity, anti-PLA2R data were concordant with the proteinuria data, and anti-PLA2R levels decreased to a greater extent and on a steeper slope in the rituximab group compared to the cyclosporine group among participants who achieved the composite outcome (Figure S10).
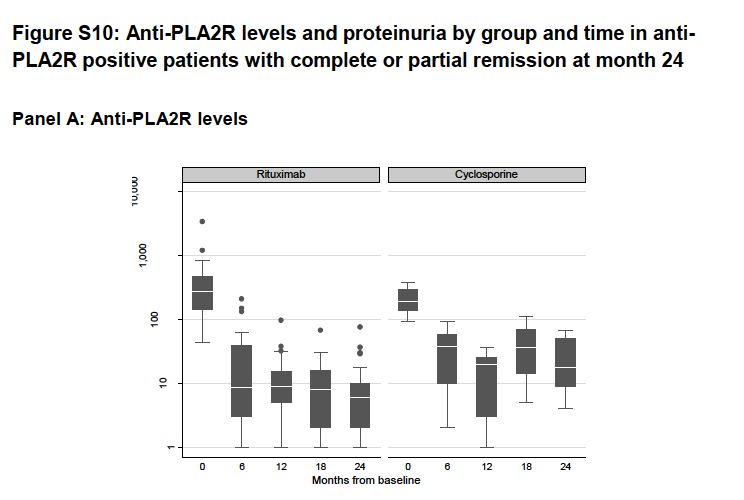
Looking at the other side of the coin, by 24 months a greater proportion of patients had treatment failure in the cyclosporine group (80%) compared to the rituximab group (40%), with time to treatment failure plotted in Figure 2 below.
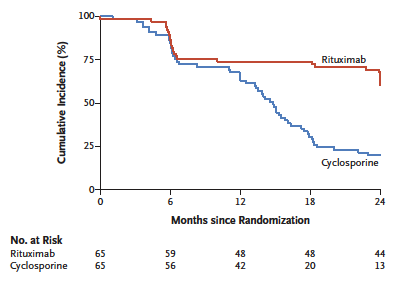
Figure 2. Time to treatment failure.
In terms of adverse events, the incidence was similar between treatment arms, although the adverse events for rituximab were infusion-related symptoms, compared to elevations in serum creatinine and gastrointestinal symptoms in the cyclosporine group (Table 3). Finally, for quality of life metrics, little difference was seen between groups.
Discussion
In patients with nephrotic-range proteinuria while on conservative therapy, rituximab was non inferior at 12 months and superior at 24 months to cyclosporine in terms of maintaining proteinuria remission up to 24 months. In short, the trial was adequately powered with a clear design that seems to circumvent the timing limitations of GEMRITUX.
Limitations and unanswered questions to consider:
The trial was not blinded to the patients and treating physicians, although objective laboratory data that formed the primary outcome were blinded to those measuring and analyzing.
Would the risk difference change if the non-responders in both arms at 6 months had continued with therapy? Would the cyclosporine arm have had more remission cases with that subgroup? What was happening to anti-PLA2R levels in non-responders?
Would we have seen fewer treatment failures (serum creatinine, adverse events requiring discontinuation) in the CNI arm if tacrolimus had been used instead?
Would we see the same results in patients of black, Asian, or Hispanic ethnicities?
How was the “positive” threshold for anti-PLA2R determined, and for future trials is there a standardized approach to measuring this analyte?
Are we missing differences among responders, non-responders, and treatment failures in terms of subtype of PMN and potentially even the different antigen epitopes of PLA2R and THSD7A?
Given the results of this trial, a portion of the nephrology community might consider moving on from CNIs and use rituximab as initial therapy. For some nephrologists, the superiority in maintaining remission would be a reason to switch to B-cell strategies. For patients, temporary adverse effects related to the infusion might be more tolerable than daily gastrointestinal upset or worry about CNI-induced nephrotoxicity. Do the results of MENTOR provide enough of an impetus to initiate immunosuppressive therapy earlier, or perhaps even in patients with disease but who are still hovering in the sub-nephrotic range, patients in whom the risk/benefit ratio of cyclophosphamide was suboptimal?
For KDIGO GN to release new guidelines, anticipated results of the STARMEN trial (tacrolimus-rituximab vs methylprednisolone-cyclophosphamide) and RI-CYCLO (rituximab vs steroids and cyclophosphamide) might need to be considered first.
This study does not provide data to make decisions about rituximab vs the modified Ponticelli protocol.
Despite some limitations and open-ended questions, the MENTOR study does seem to provide some momentum to steer the nephrology community in a B-cell centric direction for approaching PMN therapy. However, our work is not done. At the moment, one could view our available therapies as relatively blunt tools, with rituximab being an improvement but still not as sophisticated and precise as it should be.
Work remains to be completed on understanding the different subtypes of PMN further for precision medicine, linking the genetic and environmental risk factors that may trigger PMN, and engineering more specific therapies so that not all B-cells are necessarily depleted while remaining plasma cells (not expressing CD20 and thus not depleted by rituximab) producing anti-PLA2R can also be targeted. Certainly the past decade has been exciting in terms of translational developments for PMN, kick-started with the identification of PLA2R as a causal glomerular in situ antigen, and it will be even more exciting to see what these upcoming trials and future research will bring in advancing the field.
Summary prepared by Jennie Lin, Northwestern University.
Pathology slides contributed by Tiffany Caza, Arkana Laboratory